
细胞毒剂按其作用部位可分为作用于DNA、RNA的合成,作用于蛋白质合成和激素几大类。作用于DNA的抗癌药根据其作用于新合成de novo DNA,还是作用于存在的功能化(performance)DNA,又可分为DNA合成抑制剂,DNA烷化剂和DNA嵌入剂。DNA合成抑制剂多属抗代谢药物,它们的受体大都是核苷合成过程的各种酶系。DNA烷化剂和DNA嵌入剂药物的受体则是DNA本身。最近有证据证实,DNA嵌入剂和依托泊苷,替尼泊苷的受体是DNA拓扑异构酶Ⅱ,喜树碱和它的衍生物,伊立替康(irinotecam,CPTⅡ)和托泊替康(topotecan,TPT)是拓扑异构酶Ⅰ的抑制剂。由于DNA烷化剂和DNA嵌入剂能直接破坏DNA的结构及功能,故这类药物都有致癌性,它们都是周期非特异性药物。激素药的受体是胞质中的激素受体。长春新碱、秋水仙碱和高三尖杉酯碱等生物碱有抑制蛋白质合成或微管功能,紫杉醇类则是聚合的微丝蛋白的稳定剂阻断它的解聚。
常用抗癌药物按其作用于肿瘤细胞的不同周期,可以分为细胞周期特异(cell cycle specific,CCS)和细胞周期非特异(cell cycle nonspecific,CCNS)药物;按照药物的作用性质,则可分为细胞毒剂、分化诱导剂和生物反应调节剂3种。这样的分类方法对临床用药和设计联合化疗方案等都有很大指导意义。
周期特异抗癌药包括抗代谢药,如氮杂胞苷(AzCR)、阿糖胞苷(Ara-C)、氟尿嘧啶(5-FU)、6-巯基嘌呤(6-MP)、甲氨蝶呤(MTX)和6-硫鸟代嘌呤(6-TG)等,多肽抗生素——博来霉素(BLM)和生物碱类如依托泊苷(VP16)、替尼泊苷(VM26)、长春碱(VLB)、长春新碱(VCR)和长春酰胺(VDS)等。周期特异抗癌药仅能杀灭细胞增殖周期中某一期的细胞。现已证实,周期特异抗癌药对血液肿瘤和大部分处于增殖期的肿瘤是有效的。
周期非特异抗癌药包括烷化剂,如白消安(BUS)、环磷酰胺(CTX)、美法仑(MEL)和苯丁酸氮芥(CB 1348);抗生素类,如柔红霉素(DNR)、多柔比星(ADM)、丝裂霉素(MMC)等;生物碱类,如高三尖杉酯碱(HHT)、喜树碱(CPT)等,其他如顺铂(CCDP)、亚硝脲(HU)、胺吖啶(AMSA)、米托蒽醌(MTZ)等几类抗癌药。周期非特异抗癌药则对增殖周期中各期细胞均有杀灭能力,对低生长实体瘤也有作用。
当使用周期特异药物治疗时,肿瘤干细胞必须也处于增殖期而不是G0期,这是联合化疗方案设计的主要根据之一。对急性白血病等倍增迅速的肿瘤,可先用周期特异药(如Ara-C或VCR)直接杀灭处于S期的细胞,再用周期非特异药(如DNR)以杀灭残留白血病细胞,可收到协同杀灭作用,以提高化疗效果。但DNR也能杀灭S期细胞。
抗癌药物作用机制主要是药物作用于靶点也即广义受体,它们包括:①膜受体:EGFR,HER2,CD19,CD20,Pgp,KDR等。②信号分子:酪氨酸激酶(tyrosine kinase),ras蛋白,法呢基转移酶(Farnesyl transferase),蛋白激酶C(protein kinase C,PKC),丝裂原活化的蛋白激酶(mitogen activated protein kinase,MAPK),激活它的激酶MAPK/ERK Kinase,MAPKK。
其作用模式如下:刺激信号-MAPKK-MAPK-C-jun,C-fas-转录-基因表达-生物效应。①酶:二氢叶酸还原酶(dihydro folic acid reductase,DHFR);核苷酸还原酶(ribonucleotide diphosphate reductase,RDR);DNA聚合酶(DNA polymerase);RNA聚合酶(RNA polymerase);胸苷酸合成酶(thymidylate synthetase);DNA拓扑异构酶Ⅰ和Ⅱ,(DNA topoisomeraseⅠ,TopⅠ&TopⅡ);细胞周期性蛋白依赖激酶(cyclin dependent kinase,CdK);端粒酶(telomerase)。②核受体:雌激素受体(ER),糖皮质激素受体(PR),维A酸受体(RA)。③核酸:RNA,DNA。④蛋白:微丝蛋白。⑤基因:p53,bcl-2,Abl等。
现将常用细胞毒剂的分类与作用机制归纳在下表,并简要介绍如下。
细胞毒剂分类及其作用机制
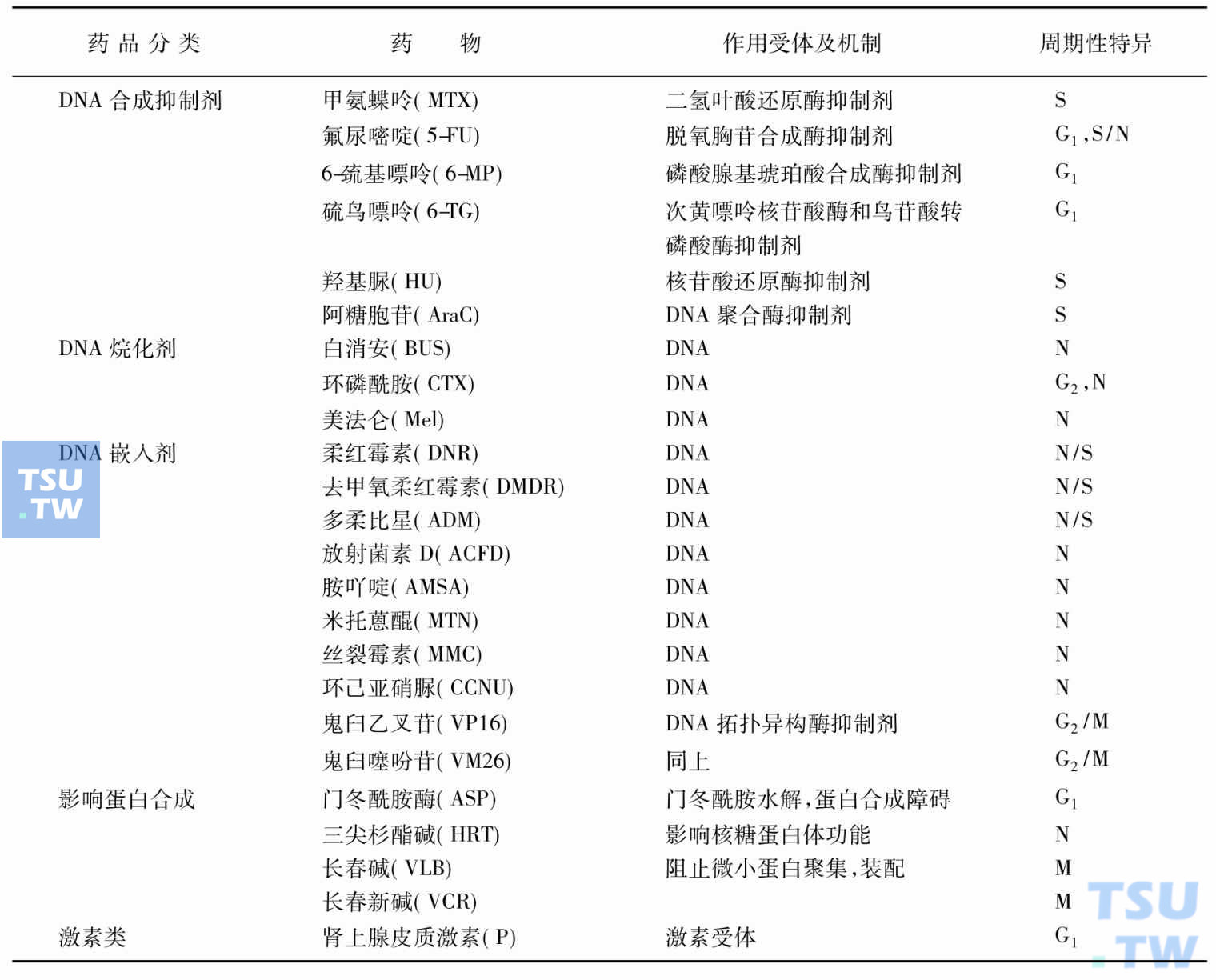
作用于DNA的药物
DNA合成抑制剂
一、甲氨蝶呤(methotrexate,MTX)
在DNA合成抑制剂中,对甲氨蝶呤作用机制的研究进展最大,现已明确其受体是二氢叶酸还原酶(dihydrofolate reductase,DHFA),已分离得到该酶的结晶,同时也搞清了它的氨基酸组成和顺序,还用X光衍射结合分子图像方法建立了二氢叶酸还原酶立体分子图像,使甲氨蝶呤与之结合的情况可用立体透视图像显示出来,从而在分子水平上阐明二者的结合区域的立体配置、疏水区域、电性相互作用强度与距离,并为计算机设计新的二氢叶酸还原酶抑制剂提供一个分子模型(下图)。
甲氨蝶呤是二氢叶酸还原酶的抑制剂。后者催化二氢叶酸成为N5,10-甲基四氢叶酸(N5,10-CH2FH4)。这是嘌呤和嘧啶在生物合成过程中的关键化合物,它使脱氧尿苷酸(dUMP)甲基化合成脱氧胸苷酸(dTMP)。甲氨蝶呤与二氢叶酸还原酶结合比它的底物二氢叶酸强1000倍,其原因可从它们的分子比较中看出它们的唯一差别在二氢叶酸4位的羟基上。甲氨蝶呤分子与之对应的4位上是氨基,同时在N10位上多一个甲基。由于利用生物同电异素原理,由氨基取代羟基,使甲氨蝶呤分子头部蝶呤环碱性增加3个pK单位,使它在生理pH下,1位氮N1质子化比二氢叶酸中不能质子化的N1与酶活性中心Asp-27的羧基结合更强。一旦结合,蝶呤环在结合区域内电子发生重排,从而提供结合时所需的能量。
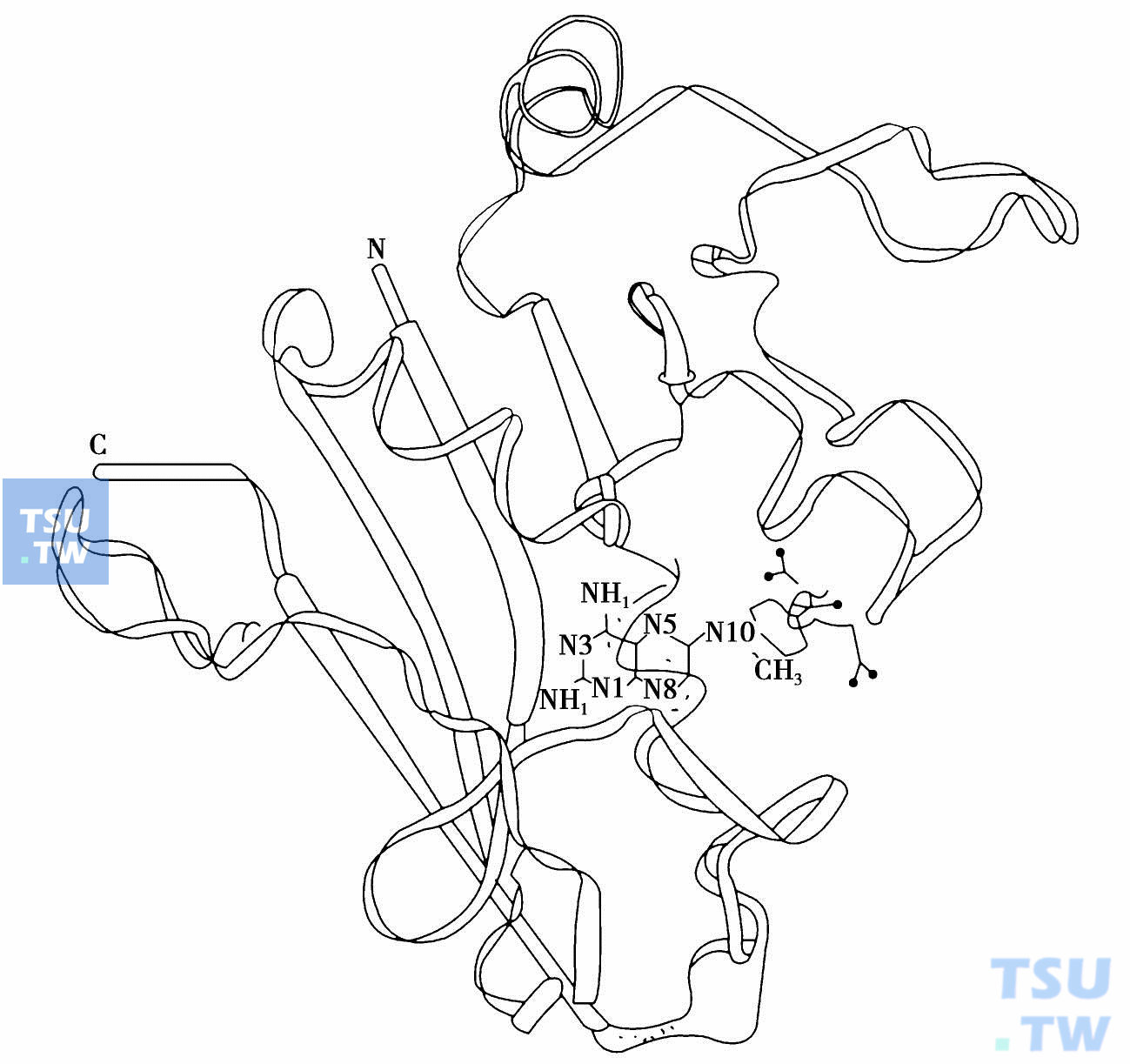
甲氨蝶呤与二氢叶酸还原酶结合模拟图
甲氨蝶呤的救援剂为甲酰四氢叶酸(醛氢叶酸,CF)和5-甲基四氢叶酸(5-MFH4)。醛氢叶酸在细胞内可产生四氢叶酸,5-甲基四氢叶酸在正常细胞内易受酶的作用转变为四氢叶酸,而在肿瘤组织中此反应较慢,故可在保持甲氨蝶呤对肿瘤细胞毒性的同时,降低其对正常细胞的毒性而达解救之目的。其救援机制可用下图表示。
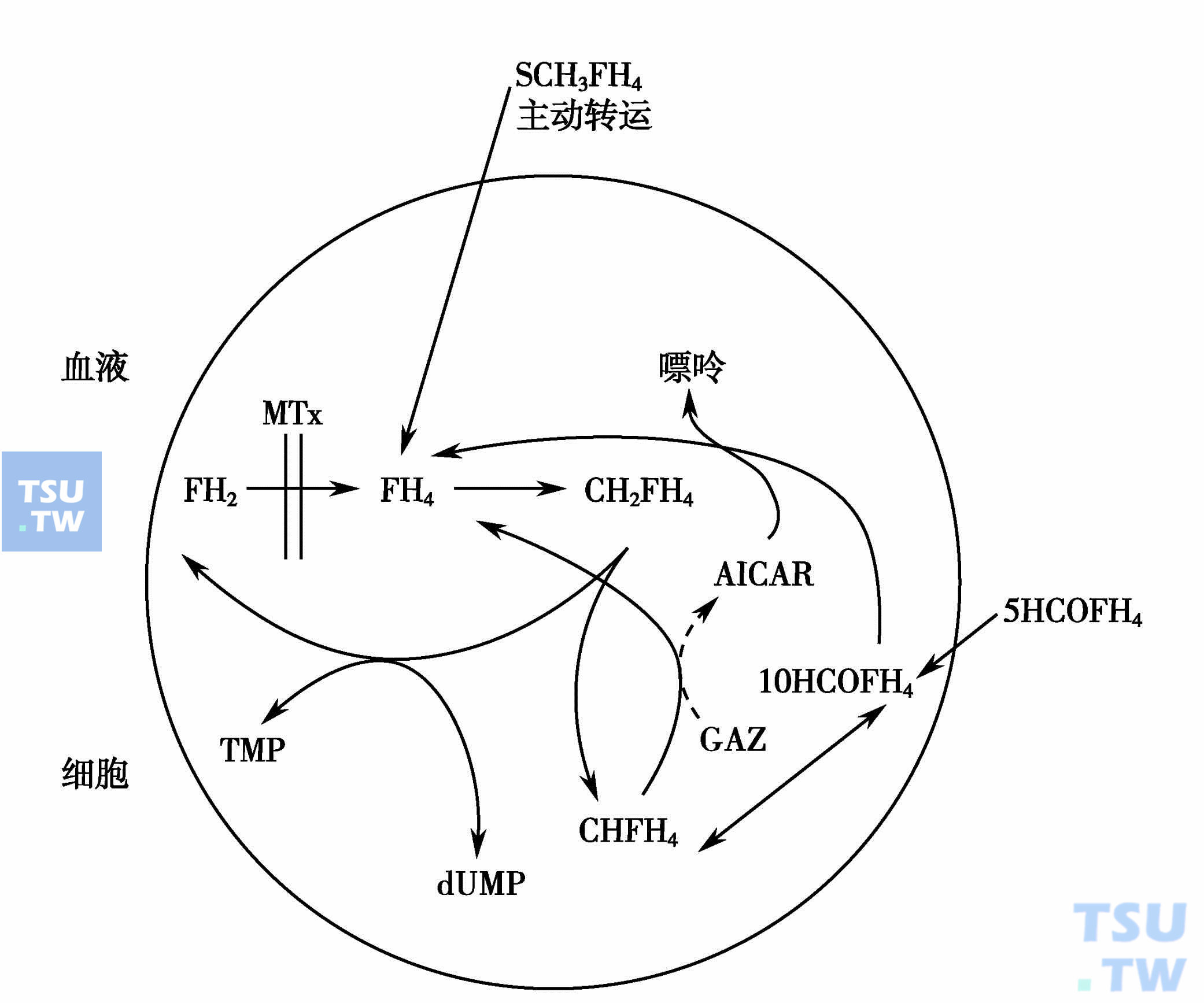
甲氨蝶呤的救援剂为甲酰四氢叶酸(醛氨叶酸,CF)和5-甲基四氢叶酸(5-MFH4)作用模式
二、嘌呤核苷酸合成抑制剂
这类药物包括6-巯嘌呤(6-MP)、溶癌呤(AT-1438)、硫鸟嘌呤(6-TG)和硫唑嘌呤等。这类药物的结构都是以相应的腺嘌呤、鸟嘌呤或次黄嘌呤为母核,应用生物同电异素原理,以巯基SH或其衍生物取代腺嘌呤4位氨基或鸟嘌呤4位羟基而成的酶抑制剂。6-巯嘌呤的作用机制,现认为是先与次黄嘌呤竞争次黄嘌呤核苷焦磷酸酶,使其本身转为活性的硫代次黄核苷酸或硫代肌苷酸(thioinosine,TIMP),同时也可阻止次黄嘌呤变成次黄嘌呤核苷酸(肌苷酸,IMP)而被利用。硫代肌苷酸的作用是:
- 抑制磷酸腺苷琥珀酸合成酶,阻止肌苷酸变成磷酸腺苷琥珀酸(AMPS),还阻止后者进一步转变为腺苷酸(AMP),最终干扰核酸合成,故用三磷腺苷(ATP)可抵消6-MP的作用。
- 阻止肌苷酸脱氢酶使肌苷酸不能转变为黄嘌呤核苷酸(XMP),从而不能生成鸟苷酸(GMP)。
- 硫代肌苷酸进一步变成二磷酸及三磷酸硫代肌苷(TIDP及TITP),后者可抑制ATP变成辅酶Ⅰ及Ⅱ(NAD及NADP),影响辅酶的功能。
- 硫代肌苷酸也可转变成硫代三磷酸鸟苷(67GTP)后掺入DNA中,阻止DNA与RNA合成。
三、DNA多聚酶抑制剂
本类药物包括阿糖胞苷(cytarabine,Ara-C)、环胞苷(Cyto cytidine,Cydo C)、氟环胞苷(AAFC)和氮杂胞苷(SA2 CR)。阿糖胞苷在体内经脱氧胞苷激酶催化磷酸化,成为三磷酸阿糖胞苷(Ara-CTP)活性代谢物才起作用。后者与天然底物dCTP竞争与DNA聚合酶结合,是DNA聚合酶的抑制剂。由于在白血病细胞和淋巴组织中脱氧胞苷激酶的含量较高,故对白血病细胞呈现一定的选择性。在敏感白血病细胞中,三磷酸阿糖胞苷浓度较高,维持较久,对S期细胞抑制明显。
最近研究证实,阿糖胞苷掺入DNA也是它的细胞毒性之一。在人早幼粒白血病细胞株和急性髓细胞白血病患者的早幼粒细胞中,阿糖胞苷是直接掺入DNA的,掺入的量与这些细胞失去克隆性相关。此外,阿糖胞苷掺入DNA还导致染色体形态异常和DNA损伤修复能力降低;其诱导染色体损伤的程度与药物接触时细胞所处周期有关。
四、核苷酸还原酶抑制剂
核苷酸还原酶(RR)是将核苷转化成脱氧核苷,即DNA合成和细胞复制限速反应的关键酶,该酶的活性和细胞复制密切相关。生长缓慢的细胞该酶的活性低,生长迅速或恶性肿瘤细胞该酶的活性明显升高。脱氧核苷池很小,故不能长久支持DNA合成,它们须由参入DNA前合成出来。因此,它是一个很好的药物靶子。
核苷酸还原酶抑制剂用于临床者有羟基脲(hydroxyurea,HU)、肌苷二醛(inosine dialdehyde)、腺苷二醛(adenosine dialdehyde)、羟基胍(hydroxy guanidine,HG)和胍那唑(guanazole,GZ)等。羟基脲是此类药的代表。它能抑制核糖核酸还原成脱氧核糖核酸,羟基脲的主要作用方式是抑制核苷酸还原酶,可能通过与核苷酸还原酶辅酶Fe2+形成螯合物使其失去活性。羟基脲对S期细胞有选择性杀伤作用,属细胞周期特异药物。由于它能使G1/S的过渡发生停滞,可用于细胞同步化,使细胞集中于G1期,从而增加放射治疗的敏感性。
DNA烷化剂
(alkylating agent)又称烷化剂,是一类化学性质很活泼的化合物,能与多种化合物,特别是细胞成分中的功能基因发生烃基反应。即用其本身在体内形成正碳离子取代生物物质中的活性氢或带有未结合电子对基因(亲和基因)形成共价结合。核酸、蛋白质或酶等分子中的羟基、巯基、胺基、羧基、磷酸基等都是亲核基团,易与烷化剂反应。烷化剂的每个分子可有1至多个烃化基因。
烷化剂作用机制可用下图来表示。
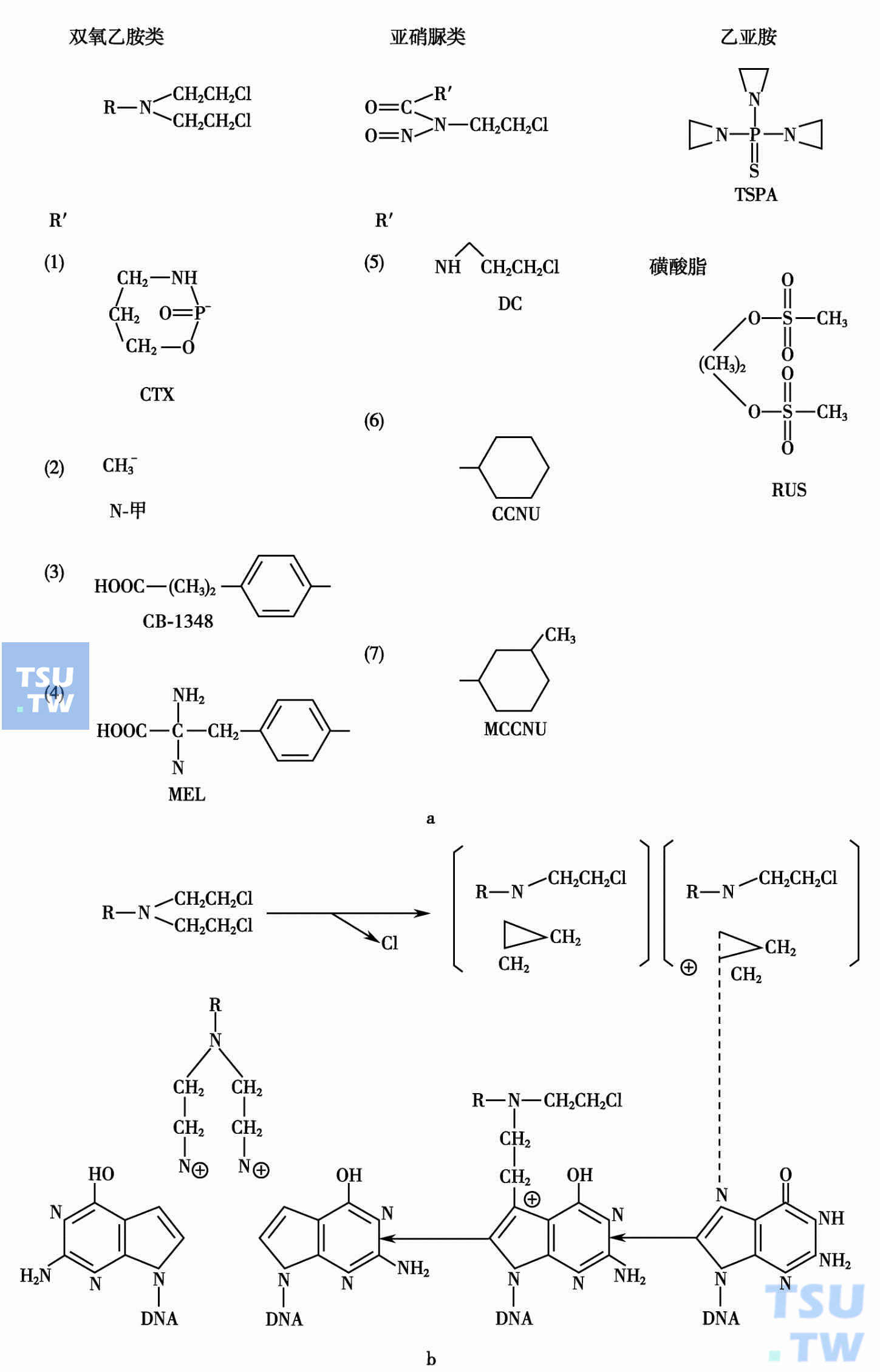
熔化剂作用机制
一个双功能的烷化剂在体内脱去氯离子后,生成一个阳碳离子,它与DNA的一个碱基产生烃基化的嘌呤,进一步反应使DNA联结。交叉联结DNA易于断裂而致细胞死亡。DNA的损害在有些细胞可以修复,可产生耐药性。
事实上,DNA的结构改变之后,其模板功能受到破坏,DNA的复制和转录也必然受干扰,蛋白质合成也受影响。此外,也发现部分烷化剂可浓集于生物膜,包括细胞膜与线粒体膜上等,影响氨基酸等物质的转运,干扰电子和能量的传递,抑制氧化磷酸化或使之解耦联。所以,细胞成分受到烃化作用之后,其功能改变是复杂的。
烷化剂均属细胞周期非特异药物,一般以G1后期和S期早期细胞较为敏感。环磷酰胺(cyclophosphamide,cytoxan,CTX)、异环磷酰胺(ifosfamide,IF)及氰乙环磷酰胺(trofosfamide,TFF)均属代谢活化烷化剂。本类药的氯乙胺基上的氯不易解离,未代谢前无烃化活性。实践证明,本类药依赖于肝细胞微粒体酶系统,即混合功能氧化酶系,在细胞色素P450的存在下,先经氧化产生4-羟环磷酰胺或醛磷酰胺。它们分解释放出烃化很强的氯乙基磷酰胺,即引起细胞毒的最终产物,此过程即称毒化作用。同时还释放出具有泌尿道刺激作用的丙烯醛,环磷酰胺类所以具有较高的抗癌选择性,是由于在敏感肿瘤细胞中发生毒化作用较强,而在肝肾等组织中,则4-羟环磷酰胺及醛磷酰胺分别转变为化学活动性较低的降解产物:4-酮基环磷酰胺或羟基磷酰胺,随后排泄,故对正常组织的损害较轻。还有认为4-羟环磷酰胺可与巯基化合物结合,生成活性较弱的4-巯基环磷酰胺,后者又可能再活化显效。在不同组织中变为巯基衍生物失活的程度不同,再活化的程度亦不同,因此药物对某些组织的选择性也不同。
根据环磷酰胺代谢活化途径,设计合成4-氢过氧环磷酰胺(4-hydroperoxy cyclophosphamide,4HC)和4-硫代乙碳酸环磷酰胺(AZTA E 7557),已在临床用于白血病自身骨髓移植的骨髓净化。其作用机制由于4HC分子中4-HOO极性基团很活泼,在细胞内很快转变成4-羟环磷酰胺而起作用。相似地,AZTA E转变成相应为4-SH衍生物而起作用。
环磷酰胺要求肝脏微粒体混合功能氧化酶催化激活,故抑制该酶的药物可降低其活化程度。体外实验证明,阿托品、麻黄碱、阿扑吗啡,及在较弱程度上细胞色素C和一些甾体激素可抑制环磷酰胺活化。泼尼松有即发的抑制作用,但每日用药较长期则加快活化速度。合用氯喹可增强环磷酰胺毒性。总之,多种药物可干扰环磷酰胺代谢,影响疗效或毒性,值得注意。
本类属细胞周期非特异药,主要阻断G2期细胞。
DNA嵌入剂
放射菌素D、蒽环类、胺吖啶、米托蒽醌、玫瑰树碱和喜树碱都是通过药物分子嵌入细胞的DNA,使DNA链断裂而死亡,这类药物通称DNA嵌入剂。其中最重要的是蒽环类的柔红霉素(DNR)、多柔比星(ADM)、4-去甲氧柔红霉素(DMDR)和表柔比星(EPR)等。由于蒽环类在肿瘤化疗中的重要性,对这类药物的作用机制研究也相当深入,并已取得明显进展。
用紫外位移、荧光淬灭、DNA黏度增加、沉降系数降低等指标,都能证明蒽环类抗生素可以嵌入DNA分子内,从而抑制DNA的复制和转录。为了进一步研究蒽环类药物与DNA受体结合及相互作用,阐明该类药物的构效关系。Quigley合成了与DNR互补的DNA 6个碱基对片段:d(CpGpT-pApCpG),并将此片段与柔红霉素一起孵育,生成橙红色晶体。光谱分析证明,每段6个核苷酸与柔红霉素分子之比为1:1。因此,其分子嵌入DNA双股螺旋d(CpGpTpApCpG)片段形成的复合物含有两个柔红霉素分子和90个水分子。
根据复合物结晶的X线衍射三维坐标绘制的模式图(下图)中可以看出,在图中DNR的ABCD环用黑点勾出位于纸面,相邻碱基对G2-G5*用粗线条表示位于纸面下方。右图则显示DNR嵌入DNA片段d(CpGpTpApCpG),分子间相互作用模式图。从图中可以看出,DNA分子中D环9位羟基和碱基G2上的N2和N3生成两个氢键,D环13位的羰基和碱基C1的O2通过一个水分子生成一个氢键桥。
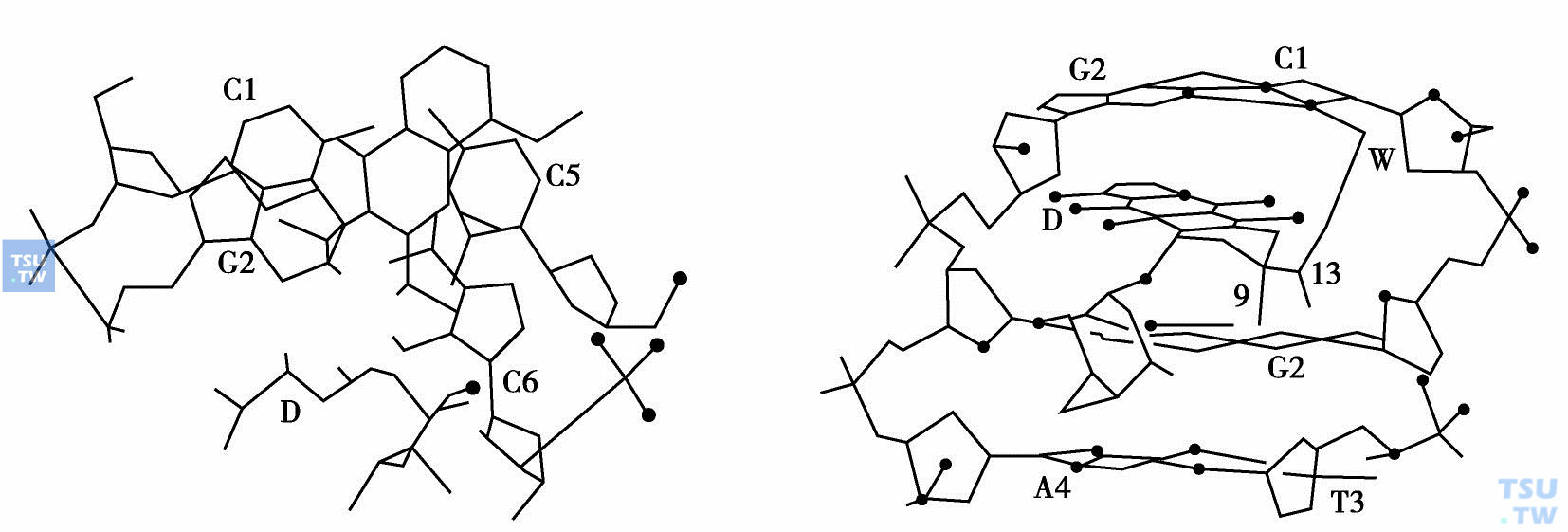
DNR与DNA结合模式图
进一步用计算机分子绘图研究复合物构象,以及与药物相互作用和构效关系。在下图中绘出了“移走”两个药物分子后d(CpGpTpApCpG)片段的分子图。由下图可以看到,DNA采取了歪扭的右手螺旋结构以适应药物分子的嵌入。中间碱基对AT的堆积方式与B-DNA类似。一般说来,嵌入剂与DNA结合时要引起DNA的解旋。在复合物DNA-d(CpGpTpApCpG)中,除了DNA分子有8°的解旋外,还可看到碱基对G2~C11向大沟(右下侧)一侧发生了移动。这种碱性基对移动主要是由于氨基糖部分位于小沟(左上侧)内,以及DNR分子的O9能与G2生成强的氢键造成的。
DNR与DNA结合计算机模拟图
DNR分子与DNA片段的相互作用主要是通过氢键和范德华力相互作用,其中氢键作用对抗癌活性是很重要的。由于O9与G2的N2、N3的距离分别为2. 91Å和2. 61Å,因而O6与N3能形成强的氢键,这就可以解释为什么C9位取代是很重要的,尤其是9位羟基取代对抗癌活性来说是必不可少的。
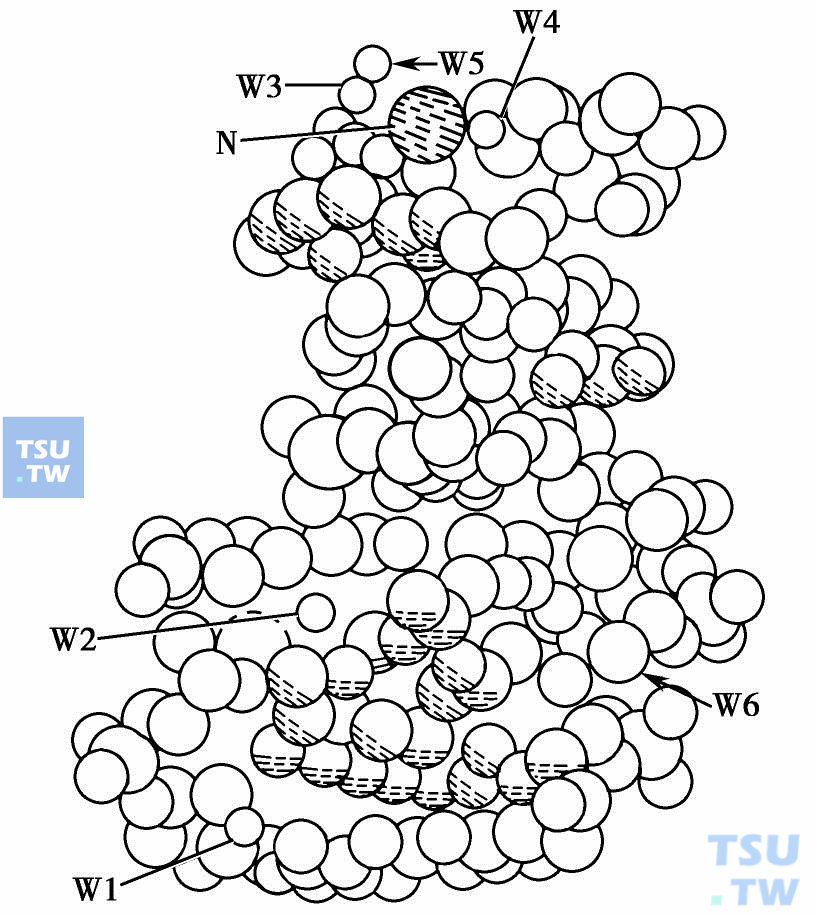
下图 将几个位置比较重要的水分子和钠离子绘了出来。DNR分子中的N3原子距离水分子W2、 W6和碱基C5的O2(或C11)较近,分别为2. 69Å、2. 80Å和2. 62Å,因此,N3能与三者形成较强的氢键。由此可见氨基N3在DNR抗癌活性的重要性。Na+能与G6的N1(2. 77Å),O4(3. 03Å),O5 (3. 10Å)和三个水分子W3、W4和W5形成配位体,所以Na+的存在增强了DNR分子与DNA的相互作用,增加了复合物的稳定性,从而影响了药物分子的生物活性。
DNA与DNR结合模型移走一分子DNR
由于蒽醌发色团平面嵌入碱基对CpG之间,因此在蒽醌发色基平面上有取代基从空间效应上考虑是不利的,环D由碱基对伸向大沟,4位的取代基越小越好,实验已证明,4-去甲氢阿霉素DMDR比DXR和DNR有更高活性。因为O14能与水分子结合形成氢键,这里的水分子又能与磷酸酯骨架形成氢键而起到桥连作用,因而14位羟基或酯基取代对提高抗癌活性是有利的,ADM便是14羟基DNR。
DNA拓扑异构酶Ⅱ(TOPO)抑制剂
新近发现,鬼臼毒素(podophyllotoxin)的抗癌作用是通过抑制DNA拓扑异构酶Ⅱ(topoisomeraseⅡ,TopoⅡ)杀灭肿瘤细胞的。进一步研究发现,上述DNA嵌入剂也是该酶的抑制剂。在增殖性肿瘤细胞中该酶的含量比正常细胞高,它是抗癌药很好的攻击靶点。尽管这些抗癌药物的作用方式是独特的,但引起DNA断裂的位置都是特异的。作为酶的抑制剂,它们和DNA、酶形成一个三元复合物,即药物-酶-DNA,而使酶-DNA复合物稳定化,使正常的酶-DNA复合物无法与ATP再结合,因而破坏DNA链的通过和复新连环。稳定化三元复合物可以用DNA损伤分析法测定DNA链断裂而得到证实。
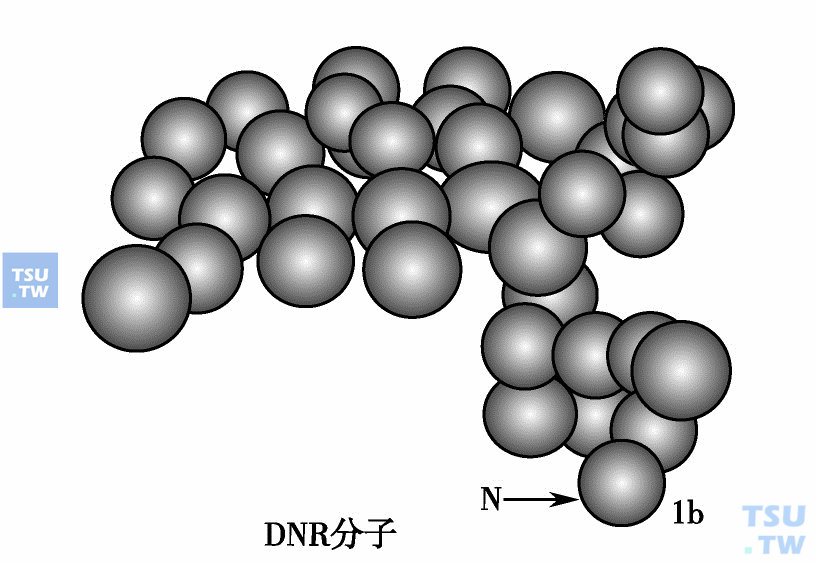
DNR分子模型
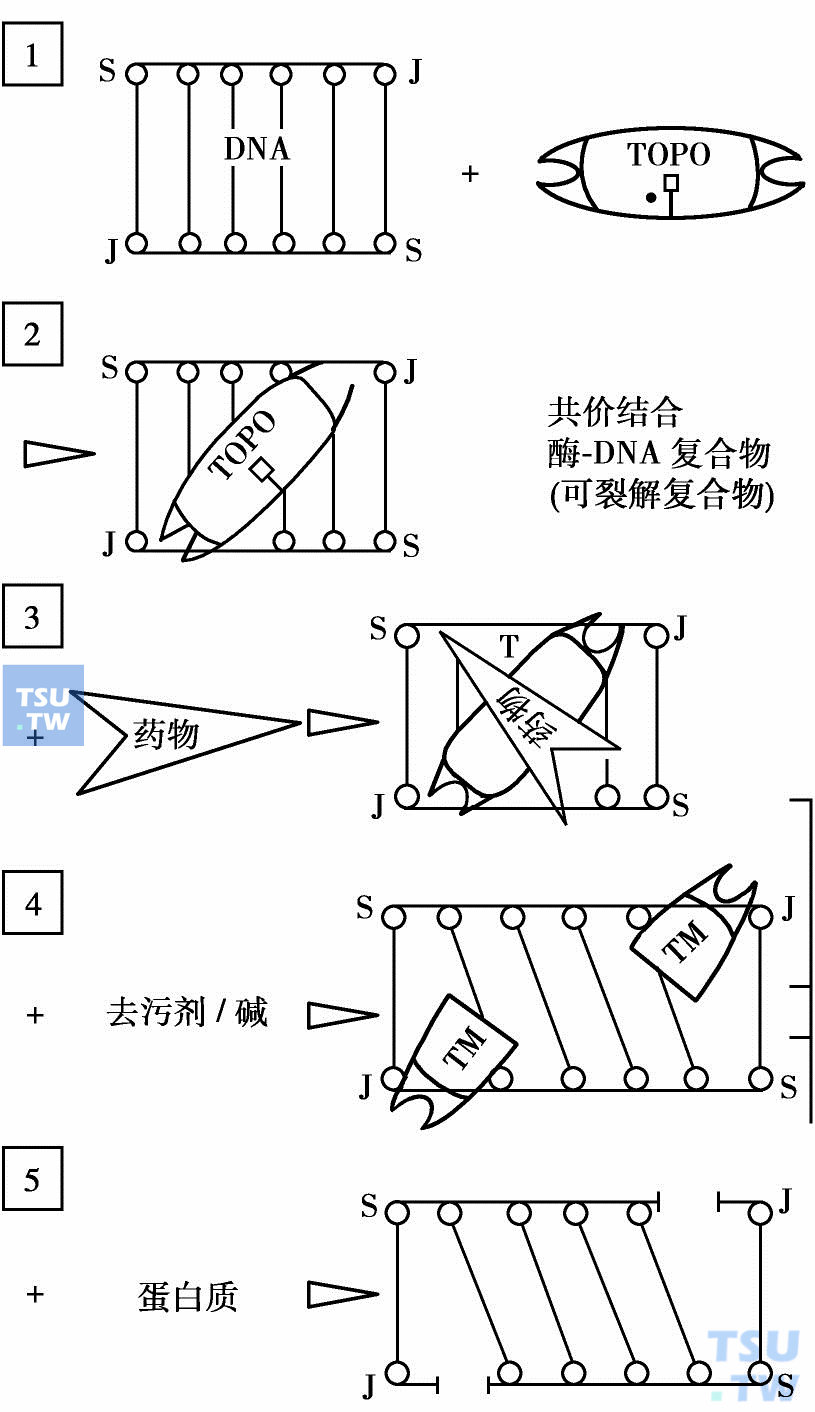
DNA TopoⅡ抑制剂作用模式图
影响蛋白质合成的药物
影响微管蛋白形成的药物
微管(microtubule)和微丝(microfilament)普遍存在于真核细胞内,参与细胞许多功能调节,包括染色体运动、细胞形状变化、内吞外排、稳定细胞表面受体,以及与绒毛、鞭毛的有关运动。在细胞内微管是处于聚合体和可溶性亚基池的动态平衡状态。实验证明,低温、高压及Mg2+可使平衡向聚合体方向移动,但Ca2+则促进微量的解离。
作用于微管蛋白的药物主要有长春碱类和秋水仙碱,其他类似作用的药物还有美登素、红豆杉醇和鬼臼毒素等。这类药物多属有丝分裂抑制剂,是作用于M期的细胞周期特异药物。
长春碱类:本类包括长春碱(vinblastin,VLB)、长春新碱(vincristin,VCR)、长春碱酰胺(vindclsin, VDS)及环氧长春新碱(formyl leurosine)。
VLB类药物影响微管装配、解聚平衡的主要原因是它们能与微管蛋白特异结合。一般认为,VLB至少可通过以下3种途径与微管蛋白作用:
- 结合于微管终端,由于空间位阻效应,阻止微管蛋白进一步聚集形成微管;
- 与微管蛋白上专一性低的亲和力位点结合,影响管蛋白自动聚集;
- 与微管蛋白上专一性的高亲和力位点结合形成微管蛋白复合物结晶。
鉴于Ca2+调素对微管蛋白和纺锤体形成具有调控作用,因此,VLB类药物对Ca2+调素的影响引起了广泛的注意。VLB、VCR和VDS对细胞膜上依赖钙调素Ca2+-ATP酶有较特异的竞争性抑制作用。
干扰核糖核蛋白体功能药物
主要有三尖杉酯碱(harringtonine)和它的异构体异三尖杉酯碱(isoharringtonine),同系物高三尖杉酯碱(homoharringto-nine)以及脱氧三尖杉酯碱(deoxyharringtonine)。
三尖杉酯碱能抑制真核细胞蛋白质合成的起始阶段。它加入网织红细胞溶解物的制备中,不久即可使聚核糖体(polyribosome)分解,释放出新生肽链,但不阻止mRNA及氨基酰tRNA与核糖体结合。超微结构表明,它们对癌细胞中与蛋白质合成有关的细胞器损伤明显,使聚核糖体解聚。近来发现,高三尖杉酯碱能抑制c-myc基因的表达,这可能是它的原发效应。本类药对G1、S和G2+M期细胞都有抑制作用,属细胞周期非特异药。
干扰氨基酸供应的药物
L-门冬酰胺酶(L-asparaginase,L-Asp)。某些肿瘤细胞,主要包括淋巴白血病细胞等,不能自行合成门冬酰胺,必须从细胞外摄取。L-Asp可将血清中门冬酰胺分解,使瘤细胞缺乏门冬酰胺,而致蛋白合成障碍,繁殖受到抑制。而正常细胞可合成门冬酰胺,受影响较小。但有些正常淋巴细胞也需要外源性门冬酰胺,因此,也会产生淋巴细胞减少的不良反应。
肾上腺皮质激素类药物
肾上腺皮质激素,特别是糖皮质激素临床用以治疗急性淋巴白血病、淋巴瘤、骨髓瘤和其他血液肿瘤。它们能促使淋巴细胞的脂肪的解体,却阻止其再酯化与利用,形成细胞内脂肪酸堆积,导致核先破裂,然后细胞解体。这种作用机制,已被新近实验证实是通过与肿瘤细胞内激素受体结合形成复合物,然后移位到核的DNA上而致的结果。并且发现敏感肿瘤细胞比正常细胞激素的受体水平高,还在这些肿瘤细胞中鉴定出一些糖皮质激素高度特异的受体。
