
蛋白质生物合成是在核糖体进行的,氨基酸按照模板mRNA的指令组装成多肽链。新生的多肽链需要折叠,并经常发生化学修饰,才能产生有活性的蛋白质分子。有活性的蛋白质分子一旦产生,某些限制其寿命的特殊的氨基酸顺序将引导它降解。
氨基酸顺序提供多肽链折叠信息
在多肽链骨架中,只有8个键角是立体化学所允许的。因此,含有n个氨基酸残基的多肽链在理论上可以产生8n种构象。然而,所有的蛋白质分子通常仅采取一种构象,即最稳定的天然构象。
天然牛胰RNA水解酶A(RNase A)分子由一条多肽链组成,含124个氨基酸残基,其中有8个Cys残基形成4对二硫键。1961年,C.Anfinsen等完成了著名的RNase A变性-复性实验。他们首先利用8mol/L尿素和β-巯基乙醇(还原二硫键)使之变性,然后去除变性因素,结果RNase A的活性完全恢复,4对二硫键准确无误地在原位重建。在8个Cys残基之间形成4对二硫键共有105种选择(形成第一、第二和第三对二硫键分别有7、5、3种选择)。如果将124个氨基酸残基的Cα原子二面角变化和侧链基团的旋转考虑在内,RNase A可能形成的构象数量将是天文数字。然而,在RNase A复性过程中仅仅选择了一种天然构象。对此,唯一合理的解释就是,多肽链的氨基酸顺序决定其三维结构,即:蛋白质分子天然构象的折叠信息存在于它的一级结构——氨基酸顺序之中。
根据“熔球(molten globule)”假说,在多肽链折叠过程中,多肽链经历了伸展(unfolding)、熔球和天然构象3个阶段。在适当条件下,伸展的多肽链围绕疏水核心凝集成中间状态“熔球”,其中有多个二级结构单元。然后,这些二级结构单元相互作用形成三级结构,折叠成蛋白质分子的天然构象。
伴侣分子促进体内蛋白质折叠
在体外,蛋白质分子折叠的效率很低,仅有少数在几分钟内完成折叠。在体内,绝大多数蛋白质分子必须迅速、正确地折叠,以免形成无功能的错误折叠蛋白质。
在细胞内,蛋白质的浓度高达100mg/ml,这种高浓度在体外通常造成蛋白质沉淀。细胞内蛋白质高效折叠的原因在于存在伴侣分子(chaperones)。伴侣分子广泛存在于细菌到哺乳动物细胞,参与蛋白质分子的一般折叠过程。伴侣分子具有ATPase活性,能够结合并稳定蛋白质分子。伴侣分子分为两种类型:①分子伴侣(molecular chaperones),结合并稳定未折叠或部分折叠的蛋白质,防止其降解;②伴侣蛋白(chaperonins),直接促使蛋白质分子折叠。
分子伴侣主要有Hsp70蛋白家族,其成员包括Hsp70(存在于细胞质和线粒体)、Bip(内质网)和DnaK(细菌伴侣分子)。Hsp70蛋白(heat shock proteins)被首次发现,是在细胞处于热休克应激状态情况下,这种蛋白质能迅速出现。Hsp70蛋白结合ATP时呈开放型(open form),其疏水性口袋结合于靶蛋白疏水区。ATP水解使Hsp70蛋白变为封闭型(close form),释放靶蛋白。当新生的多肽链正在核糖体合成的时候,分子伴侣即与之结合(下图)。
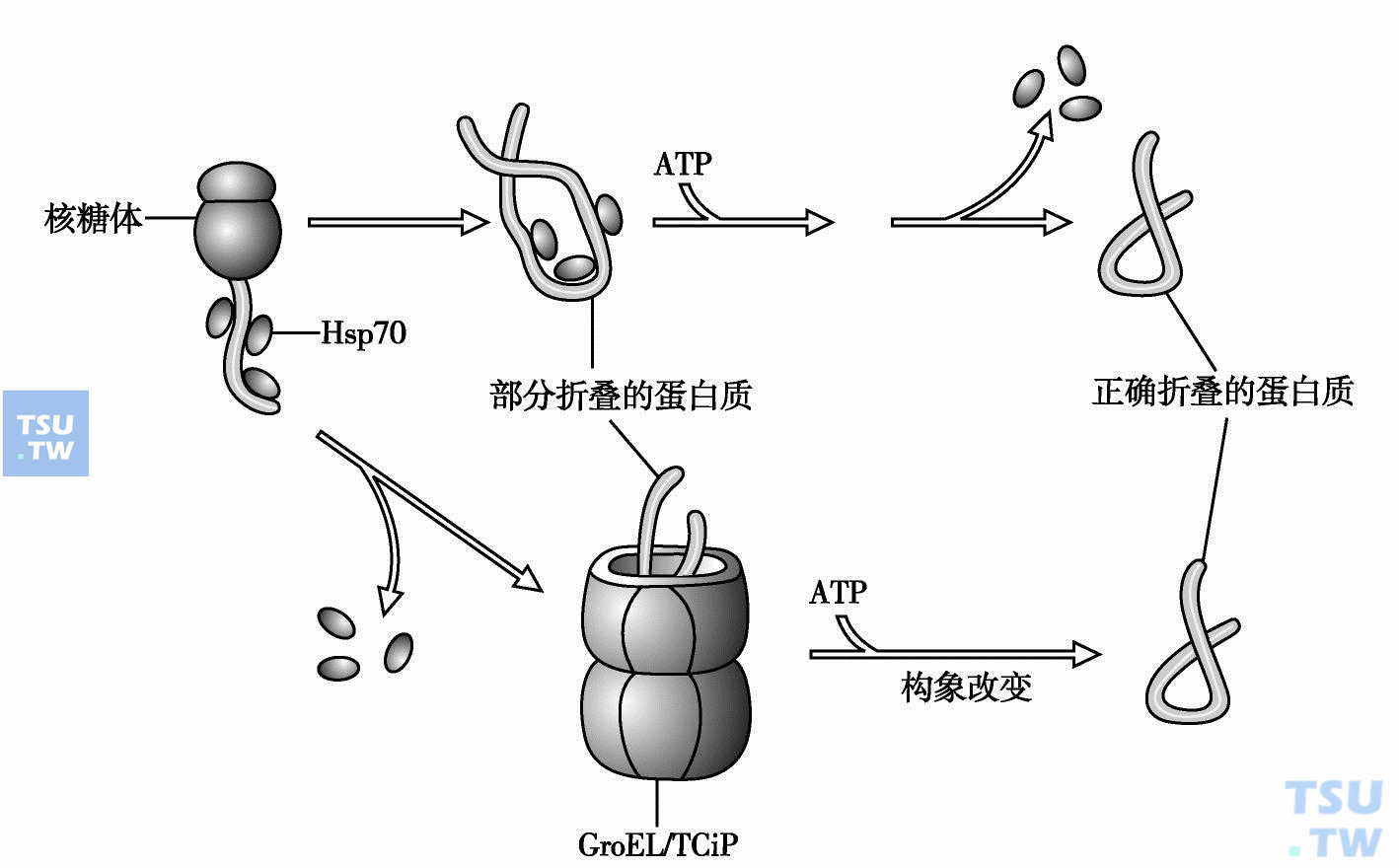
伴侣分子介导蛋白质折叠
肌动蛋白、微管蛋白等少数蛋白质的折叠需要伴侣蛋白提供帮助。真核伴侣蛋白TCiP是巨大的桶形复合物,由8个Hsp60蛋白组成,与之同源的细菌伴侣蛋白GroEL含14个相同的亚基。细菌GroEL介导的折叠机制比真核TCiP容易了解。在细菌中,部分折叠或者错误折叠的蛋白质插入GroEL的空腔并结合于空腔的内壁,进而折叠成天然构象(图8-3)。这一过程依赖ATP,需要借助于辅伴侣蛋白(co-chaperonin)GroES。真核TCiP的空腔只能允许分子量小于55kD的多肽链通过这一途径折叠。
某些蛋白质分子不需要折叠
人们曾经以为,天然蛋白质分子必须折叠成一定的三维结构才有功能。近年来发现,约30%的天然蛋白质分子完全或部分未折叠(unfolded)。完全未折叠的蛋白质被称为“天然未折叠蛋白质(natively unfolded proteins,NUP)”或“内源无结构蛋白质(intrinsically unstructured proteins,IUP)”。目前已发现约100种天然未折叠蛋白质,其多肽链长度范围50~1800个氨基酸残基,主要特征为“高净电荷、低疏水性”,含有较多的亲水性氨基酸(尤其是Lys和Glu),多Pro,而疏水性氨基酸较少,特别是芳香族氨基酸含量低。
这种缺乏特定结构的蛋白质的优势在于,具有更强的“柔韧性(malleability)”,能够结合几种不同的配体,其表面更容易进行分子间相互作用,令其功能更加多样化。
已发现的NUP(或IUP)蛋白质的功能有30多种,包括细胞信号转导、细胞周期调控、基因表达调控、DNA复制和损伤修复等。
化学修饰和加工改变蛋白质的活性
在细胞中,新生的蛋白质多肽链在核糖体合成后,其化学结构需要发生变化,以改变其活性、寿命或细胞定位。这种蛋白质化学结构变化分为两种类型,一是化学修饰(chemical modification),二是加工(processing)。化学修饰是指将化学基团共价连接到多肽链的N端、C端或内部残基侧链,往往是可逆的。加工是指去除一些肽段,一般不可逆。
多肽链N端氨基的乙酰化(acetylation)是最普遍的化学修饰方式。非乙酰化的蛋白质往往被细胞中的蛋白酶迅速降解。膜蛋白的末端及其附近残基经常加上了长长的脂质类基团,将蛋白质锚定于膜磷脂双层。
蛋白质分子内部残基的侧链可以被多种化学基团修饰,其中最重要的是Ser、Thr、Tyr的羟基发生可逆磷酸化。许多分泌蛋白和膜蛋白的Asn、Ser、Thr侧链可能成为糖基化(glycosylation)位点,结合线性或分支糖链。
蛋白酶解(proteolysis)利用蛋白酶切除新生的蛋白质的部分肽段,是一种最普通的加工方式,如胃、胰腺分泌的消化酶酶原以及凝血因子的激活。蛋白酶解也可以产生有活性的肽激素,如EGF和胰岛素。在细菌和低等真核生物中还存在一种特殊的蛋白质自我剪接(self-splicing)加工方式,切除多肽链内部的蛋白内含肽(intein),将两侧的蛋白外显肽(extein)连接起来,类似于真核mRNA前体剪接加工。
细胞蛋白质的降解途径
体内蛋白质降解分为细胞外、内两种途径。
细胞外降解途径,主要用于蛋白酶在胃、肠将食物蛋白质消化。这些蛋白酶包括:胰蛋白酶、胰凝乳蛋白酶等内切蛋白酶(endoproteases),氨肽酶(aminopeptidases,从N端依次切除残基)、羧肽酶(carboxypeptidases,从C端依次切除残基)等外肽酶(exopeptidases),以及将寡肽(oligopeptides)切割成2肽、3肽、氨基酸的肽酶(peptidases)。
细胞内有几条蛋白酶解途径,以降解错误折叠或变性蛋白质、浓度减少的正常蛋白质和外源蛋白质。途径之一是利用溶酶体(lysosomes),溶酶体是一种细胞器,内部呈酸性,含有大量蛋白酶。另一主要途径是泛素介导的蛋白质降解,泛素(ubiquitin)是一种含76个氨基酸残基的蛋白质。这一途径分为两步。首先,借助3种酶(E1~E3,即泛素激活酶、泛素缀合酶、泛素连接酶),使多个泛素分子共价结合于靶蛋白内部的Lys侧链。然后,泛素化的蛋白质被蛋白酶体(proteasome)降解。蛋白酶体是一个巨大的圆柱形口袋状多亚基复合物,内含多种蛋白酶,依赖ATP将泛素标记的蛋白质切割成小肽。由泛素介导降解的蛋白质分子含有降解信号,例如有丝分裂细胞周期蛋白的内部序列Arg-X-X-Leu-Gly-X-Ile-Gly-Asx,或富含Pro、Glu、Ser和Thr的内部序列(PEST序列),能够被泛素缀合酶识别。
许多细胞质蛋白质的寿命和N端残基的特性相关,提示N端残基与蛋白质泛素化有关。在体内,寿命短的蛋白质3分钟内被降解,其N端通常为Arg、Lys、Phe、Leu或Trp。而寿命长达30个小时以上的蛋白质,其N端一般为Cys、Ala、Ser、Thr、Gly、Val或Met。所有新合成的蛋白质的N端均为Met,有稳定作用,需要通过酶促反应将其切除,才能在N端产生一个去稳定作用(destabilizing)的残基。
蛋白质折叠异常可能导致疾病
某些蛋白质可能折叠成与其天然构象不同的三维结构,不仅导致失去正常功能,而且可能成为蛋白酶解的标记,而蛋白酶降解片段的积累可能引起退行性疾病(degenerative diseases),在脑、肝等组织细胞中出现不溶性蛋白质斑块(plaques)。
例如,在阿尔茨海默病(Alzheimer disease)患者脑组织中,出现不溶性蛋白质斑和纤维缠结(tangles),在原子能显微镜下可以看到许多短纤维有规律地排列。每根纤维由47个氨基酸残基组成,名为β-淀粉样肽(β-amyloid peptide)。这些纤维是大量天然蛋白质的蛋白酶解产物,包括淀粉样前体蛋白(amyloid precursor protein)和Tau蛋白(一种微管结合蛋白)。在其他器官中形成的斑块也来自天然蛋白质的蛋白酶解产物,如凝溶胶蛋白(gelsolin,肌动蛋白结合蛋白)和血清白蛋白。这些蛋白酶解产生的多肽片段发生聚合,形成非常稳定的纤维。
错误折叠蛋白质可能是哺乳动物若干脑退行性疾病的病因,其中了解得比较清楚的有疯牛病(mad cow disease)、人库鲁病(kuru disease)、克-雅病(Creutzfeldt-Jakob disease)、羊瘙痒症(scrapie)等。这些疾病可称为海绵样脑病(spongiform encephalopathies),因为患者大脑皮层细胞中充满海绵样空洞,其典型症状是痴呆、动作失调。
20世纪60年代,人们发现这种疾病的致病因子可能没有核酸,T.Alper认为这种特殊的致病因子可能是蛋白质。当时已知的致病因子病毒、细菌、真菌等都含有核酸,其毒性与基因复制和增殖有关。经过30多年的研究,S.Prusiner等发现,这种疾病的感染因子是一种蛋白质(28kD),将其命名为“prion protein(朊病毒蛋白,PrP)”。PrP是所有哺乳动物脑组织中的正常组分,功能尚不清楚。缺失PrP基因(当然朊病毒蛋白本身也缺失)的小鼠看上去正常,症状仅仅出现在正常的PrP蛋白(PrPC)的构象改变成PrPSc之时(Sc取自scrapie)。PrPSc蛋白可以将PrPC蛋白转变成PrPSc,并产生多米诺效应,将越来越多正常的PrP蛋白转变成致病型PrPSc蛋白。PrPSc导致海绵样脑病的机制尚不清楚。
在遗传型朊病毒疾病中,PrP编码基因突变导致一个氨基酸残基发生改变,可能造成PrPC转变成PrPSc。
(杨克恭)
