
限制性内切酶
限制性核酸内切酶简称限制酶,是一类存在于微生物中,能识别双链DNA上特定核苷酸序列,并在识别位点及其附近切割双链DNA的脱氧核糖核酸酶。限制酶的命名来自分离出该限制酶的微生物种属的缩写。每种酶都有其特定的识别序列和切割位点。如限制酶EcoRⅠ识别如下序列,并在GA之间将DNA分子切断。
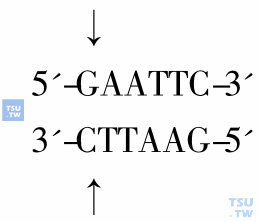
微生物种属来源不同而识别位点相同的限制酶称为同切限制酶(isoschizomer)。如HpaⅡ和MspⅠ都识别5'-GCCG-3'序列,但MspⅠ切割时不受识别序列甲基化的影响,而HpaⅡ只切割C未被甲基化的序列,不能切割C甲基化的序列。一些限制酶切割双链DNA后产生平齐的末端(blunt end),另外许多限制酶切割后在末端产生出一小段单链序列,称为黏性末端(cohesive end)。识别序列不同的限制酶可切割出相同黏性末端(compatible cohesive end)。
限制酶切割DNA的产生的片段大小,取决于该限制酶识别序列的核苷酸长度,并与被切割DNA的核苷酸组成、序列和结构特点有关。若假定DNA分子中4种脱氧核糖核苷酸的组成相等且随机排列,则限制酶切割DNA产生的片段数目及其大小,只与该限制酶识别序列的核苷酸长度有关,即被识别的序列在DNA链中出现的频率为1/4n。用识别序列为四核苷酸的限制酶切割DNA时,平均每256bp (1/44)有一个切点,或者说酶切片段的平均长度为256bp。用识别序列分别为五、六、七、八核苷酸的限制酶切割DNA时,就会依次得到平均长度分别为1024bp(1/45)、4096bp(1/46)、16 384bp(1/47)和65 536bp(1/48)的片段。在人和哺乳动物基因组DNA中,识别位点较少的限制酶称为高识别特异性限制酶或稀有切割位点限制酶(rare cutter)。
被限制酶切断的DNA片段可在DNA连接酶的作用下重新连接。这样就可以对DNA片段进行重组。另外,由于不同生物基因组的大小与核苷酸顺序的排列有很大差异,所以,不同生物种属DNA限制酶片段的长度和数目也不尽相同。因此,由限制酶片段构成的“限制酶物理图谱”可以作为不同基因组或染色体特异的结构特征。
核酸凝胶电泳
核酸凝胶电泳是使DNA或RNA分子在电场力的作用下通过凝胶,利用凝胶对不同长度核酸片段阻滞作用的差异使其分离。在电场强度设定时,所有核酸分子的电荷密度都相同,而不同长度的核酸分子在相同浓度的凝胶中迁移时受到的阻力各异,因此可以通过凝胶电泳将不同大小的核酸片段按其长度分开。选择适当的凝胶浓度可使不同范围的DNA片段得到有效的分离。提高凝胶浓度适宜分离较小的DNA片段;降低凝胶浓度可以使较大的DNA片段得到较好的分离。通常使用2%~4%的琼脂糖凝胶分离50~1500bp的DNA片段,用0. 7%~1%的琼脂糖凝胶分离1~20kb的DNA片段。但是即使使用了0. 3%~0. 5%的高强度琼脂糖凝胶也只能分离50~100kb的DNA片段,对于大于50kb的 DNA片段一般很难用普通凝胶电泳有效分离。
脉冲场凝胶电泳(pulsed field gel electrophoresis,PFGE)是使DNA分子在一个定时改变方向的脉冲电场凝胶电泳系统中泳动。由于大于30kb的DNA分子在电泳时只能以阻力最小的头尾牵引方式进入胶孔匍匐蛇行,所以每改变一次电场方向,DNA分子就要相应地重新调整一次泳动方向。长DNA片段在凝胶中移动时受到的阻力大,重新定向所需的时间长,其重新定向所需的时间与DNA片段的长度成正比。这样,DNA分子越大,它在每一个脉冲时间内用于重新定向的时间越多,净迁移的时间越少,实际泳动的距离也就越短。反复改变电场方向,并通过调节脉冲时间,就可以将各种不同长度的DNA片段有效地分开。
核酸凝胶电泳广泛用于DNA片段分离、回收、物理图谱分析与RNA鉴定。经过分离的核酸片段需用啡啶溴红(溴化乙啶,EBr)染色。在波长254nm的紫外光照射下,DNA片段显示橙红色。这是因为DNA吸收254nm的紫外光后发出次级荧光,嵌合于DNA双螺旋小沟中的EBr吸收302nm和366nm的荧光、再发出波长590nm的红色可见光。RNA分子因其具有局部二级结构,所以也可用EBr染色,但检测灵敏度大大低于DNA。由于短波长紫外光对DNA。由于短波长紫外光对DNA片段有破坏作用,所以在回收DNA片段时可使用302nm或360nm的长波长紫外光直接激发EBr,以减少紫外光对DNA片段的破坏。但这会降低检测灵敏度。
DNA修饰与合成酶
DNA聚合酶催化DNA体外合成反应。这些酶作用时大多需要模板,合成出的产物序列与模板互补。大多数聚合酶优先作用于DNA模板,也可拷贝RNA,但效率较低。最常用的以DNA为模板的DNA聚合酶是大肠杆菌(E. coli)DNA聚合酶Ⅰ及其大片段(klenow片段)、T4和T7噬菌体DNA聚合酶、测序酶(sequenase),经过修饰的T7DNA聚合酶和耐热DNA聚合酶。
E. coli DNA聚合酶Ⅰ为一分子量109kD的多肽链,具有5'→3'DNA聚合酶活性,5'→3'及3'→5'外切核酸酶活性和大肠杆菌必备的RNA酶H的活性。DNA聚合酶Ⅰ大片段(Klenow)去除了DNA聚合酶Ⅰ的5'→3'外切酶活性,只保留其5'→3'DNA聚合酶活性和3'→5'外切核酸酶活性。可用于补平DNA3'的凹端,或先利用其3'→5'外切酶活性将DNA片段的3'凸出端切成凹端,再将其补平。Klenow的这一补平作用还可用于DNA片段3'末端的放射性或非放射性标记。DNA聚合酶大片段的5'→3'DNA聚合酶活性主要用于随机引物标记DNA片段、cDNA第二链合成和双脱氧末端终止法DNA测序。
耐热DNA聚合酶是一种耐热的以DNA为模板的DNA聚合酶,分子量65kD。最初是从极度嗜热的栖热水生菌中分离出来的,其最佳作用温度为72℃。目前已从多种耐热菌中分离出耐热性更好的DNA聚合酶,广泛应用于聚合酶链反应。
以RNA为模板的DNA聚合酶称为反转录酶。常用的两种反转录酶分别来自禽成髓细胞瘤病毒(AMV)和Moloney鼠白血病病毒(Mo-MLV)。两种反转录酶在许多方面有所区别,但均无3'→5'外切酶活性。AMV反转录酶包括两条多肽链,除聚合酶活性外还具有很强的RNA酶H活性。Mo-MLV反转录酶的RNA酶H活性较弱,有利于合成较长的cDNA。AMV反转录酶的最适反应温度为42℃(鸡的正常体温),而Mo-MLV反转录酶在42℃时则迅速失活。因此,AMV反转录酶可更有效地转录具有较强二级结构的mRNA模板。二者反应时所要求的pH也有一定差异。反转录酶主要用于mRNA转录为cDNA,以便克隆、分析,或代替DNA聚合酶进行双脱氧末端终止法DNA顺序,还可用于补平5'突出的DNA片段(也可用此法标记DNA5'端)。
DNA连接酶可以催化DNA5'磷酸基与3'羟基之间形成磷酸二酯键。最常用的DNA连接酶是T4噬菌体DNA连接酶。T4噬菌体DNA连接酶为一分子量68kD的多肽。用于连接平端或末端互补的DNA分子以及DNA分子自身环化。是DNA重组中必不可少的工具酶。
基因克隆及主要的载体系统
DNA限制性内切酶、连接酶及其他各种修饰酶的使用,可以按照一定的设计将不同的DNA片段组装在一起,使其具有特定的功能;或者将某个基因或基因片段从复杂的基因组中分离出来。这一操作被称为DNA重组或基因克隆。
首先用限制酶将需要分离或重组的DNA剪切成一定长度的片段,经过一定的修饰,与载体DNA连接,形成重组DNA。然后将重组DNA送入单细胞微生物(通常为大肠杆菌或酵母)中,经过恢复培养,筛选出带重组DNA的菌落。
根据被重组DNA片段的长度和重组DNA的目的,可以选用不同的载体。常用的载体有质粒、λ噬菌体、黏粒、细菌人工染色体(bacteria artificial chromosome,BAC)、酵母人工染色体(yeast artificial chromosome,YAC)以及穿梭质粒(shuttle vector)等。
质粒是细菌中独立于细菌基因组DNA之外,能自我复制并具有稳定基因的环状双链DNA分子。天然质粒可长达数百kb,经过改造用于DNA重组的质粒一般为2~10kb。质粒中有DNA复制起始位点、外源DNA片段插入的重组位点和一定的筛选标志。将病毒DNA与质粒重组可构成既能在原核生物中扩增,又可以在真核生物中表达的穿梭质粒。含有转录起始位点等成分,可将重组DNA转录成mRNA并表达出蛋白质的质粒称为表达载体。真核细胞表达载体中除含有质粒的基本成分外,还有真核基因启动子、mRNA剪接位点、3'多聚A加尾位点以及真核细胞选择标记等。
野生型λ噬菌体DNA约48kb,其中一部分序列可被替换而不影响其裂解及生长。在λ噬菌体DNA中最多可以插入(替换)23kb外源DNA。重组DNA经噬菌体包装蛋白包装成噬菌体颗粒,以噬菌体感染的方式进入其宿主大肠杆菌,并在其中繁殖、扩增,形成蚀斑。
野生型P1噬菌体基因组约90kb,作为一个自主复制单位游离于宿主染色体之外。P1载体约30kb,在大肠菌中以质粒形式存在,含抗药性筛选标记、pBR3222质粒复制子、P1质粒复制子、P1噬菌体复制子、包装识别位点,进入宿主后重组环化的位点和一些调节重组DNA长度的填充片段。P1噬菌体载体系统可用于克隆75~110kb的外源DNA片段。
黏粒克隆是可以被包装成噬菌体颗粒的重组质粒。黏粒载体序列有5~8kb,含质粒DNA必备的复制起点、抗药性筛选标记、重组位点和将重组DNA包装成λ噬菌体颗粒所需的cos序列,因此叫做cosmid,译为黏粒或柯斯质粒。因为可被包装到λ噬菌体中的DNA片段为38~52kb,所以黏粒载体可克隆33~47kb的外源DNA片段。重组DNA经过包装,以噬菌体感染方式进入宿主菌,再环化成质粒,以质粒形式存在于宿主体内。
在质粒中克隆外源基因片段最简便,但插入片段的长度超过10kb时转化效率大大降低。λ噬菌体、黏粒和P1噬菌体克隆体系,采用将DNA包装成噬菌体颗粒后转染宿主菌的方法提高转化效率。电转化(electroporation)技术的发展为提高大质粒的转化效率创造了条件,现在已经可以构建和转化几十到二、三百kb的细菌人工染色体(BAC)克隆。
由具有完整着丝粒、端粒和自主复制顺序功能的DNA片段组成的人造酵母染色体(YAC),在酵母细胞的有丝分裂和减数分裂过程中具有与天然酵母染色体一样的稳定性。YAC载体中有酵母的着丝粒、自主复制顺序成分、可用作端粒的TEL序列,以及筛选含YAC的酵母菌株时使用的选择标记。YAC载体可插入200~1000kb的外源DNA片段,用于克隆大片段外源DNA。YAC克隆有序排列构成的物理图谱是基因克隆和DNA序列分析的基础。含完整基因位点,包括位点控制区、启动子、全部结构基因序列,表达调节信号的YAC,还可作为研究基因簇功能以及定点整合的对象,进行转基因动物实验。
分子杂交
DNA双螺旋结构是由两条序列互补的多核苷酸链接碱基配对原则以氢键结合而成。在一定条件下,如酸碱度、加热或有机溶剂的存在,可以破坏DNA双链的氢键结合将两条链分开,这一过程称为变性。变性时只破坏氢键,不涉及共价键的断裂。变性DNA两条彼此分开的链在适当条件下重新缔合成双链的过程叫做复性。在实验过程中任何两条来源不同的核苷酸链,只要各自具有一部分可以互补的序列即可形成氢链结合,产生异源双键。因此,将具有一定互补序列的核酸单链,在液相或因-液体系中按碱基酸对原则,结合成异源双链的过程称为分子杂交。DNA与DNA、DNA与RNA、RNA与RNA,以及人工合成的寡聚核苷酸链与DNA或RNA之间,只要具有一定的互补序列均可发生杂交。分子杂交可用于检测基因结构的改变、mRNA的表达、重组DNA的筛选等。被检测的核酸分子称为靶基因或靶序列;用于探测靶基因(或其表达产物)的具有一定可检测标志的已知序列称为分子探针。
探针所携带的标志物可以是放射性同位素(3H、14C、35S、32P、33P、125I等),也可以是非放射性的生物素、地高辛等半抗原类物质。探针与靶基因均在液相时称为液相杂交;靶基因在固相支持物如硝酸纤维素或尼龙膜、凝胶、载玻片上,探针在液相时称为固相杂交。通过凝胶电泳将DNA或RNA分子按片段长度分开,并转移到尼龙或硝酸纤维素膜上进行杂交叫做DNA印迹(Southern)杂交或RNA印迹(Northern)杂交。将被测核酸分子(有时是细胞裂解液)点在尼龙或硝酸纤维素膜上进行杂交叫斑点杂交。探针分子固定在膜上,靶基因在液相中的杂交称为反向杂交。在细胞或组织切片上进行的杂交称为(细胞或组织切片)原位杂交。将菌落或噬菌斑转移至尼龙或硝酸纤维素膜上进行杂交叫做菌落或噬菌斑原位杂交。
PCR技术
DNA聚合酶链反应(polymerase chain reaction,PCR)是模拟天然过程的DNA体外复制方法。在DNA聚合酶作用下,以DNA为模板,使位于模板两侧的一对寡核苷酸引物之间的序列扩增。PCR包括:变性、退火和链延伸3个步骤的多次循环。变性时通过加热破坏DNA模板双链间的氢链,使互补链分开;退火是将温度降至一定程度,使引物与模板DNA按碱基配对原则互补结合;然后在DNA聚合酶作用下,从引物的3'末端开始合成模板DNA的互补链。从第2个循环开始,前一次PCR的产物也可作为模板,指导下一次的DNA合成。理论上,每个循环后产物增加一倍。30次之后模板将被复制10亿次。由于实际上每个循环的扩增效率达不到100%,所以当使用1μg DNA为模板扩增单拷贝基因时(105模板),通常需要进行25~35个循环,才能使DNA片段的总数增加到1012左右(一般凝胶电泳下肉眼可见水平)。PCR是自动化连续进行的,所以必须使用耐热DNA聚合酶。
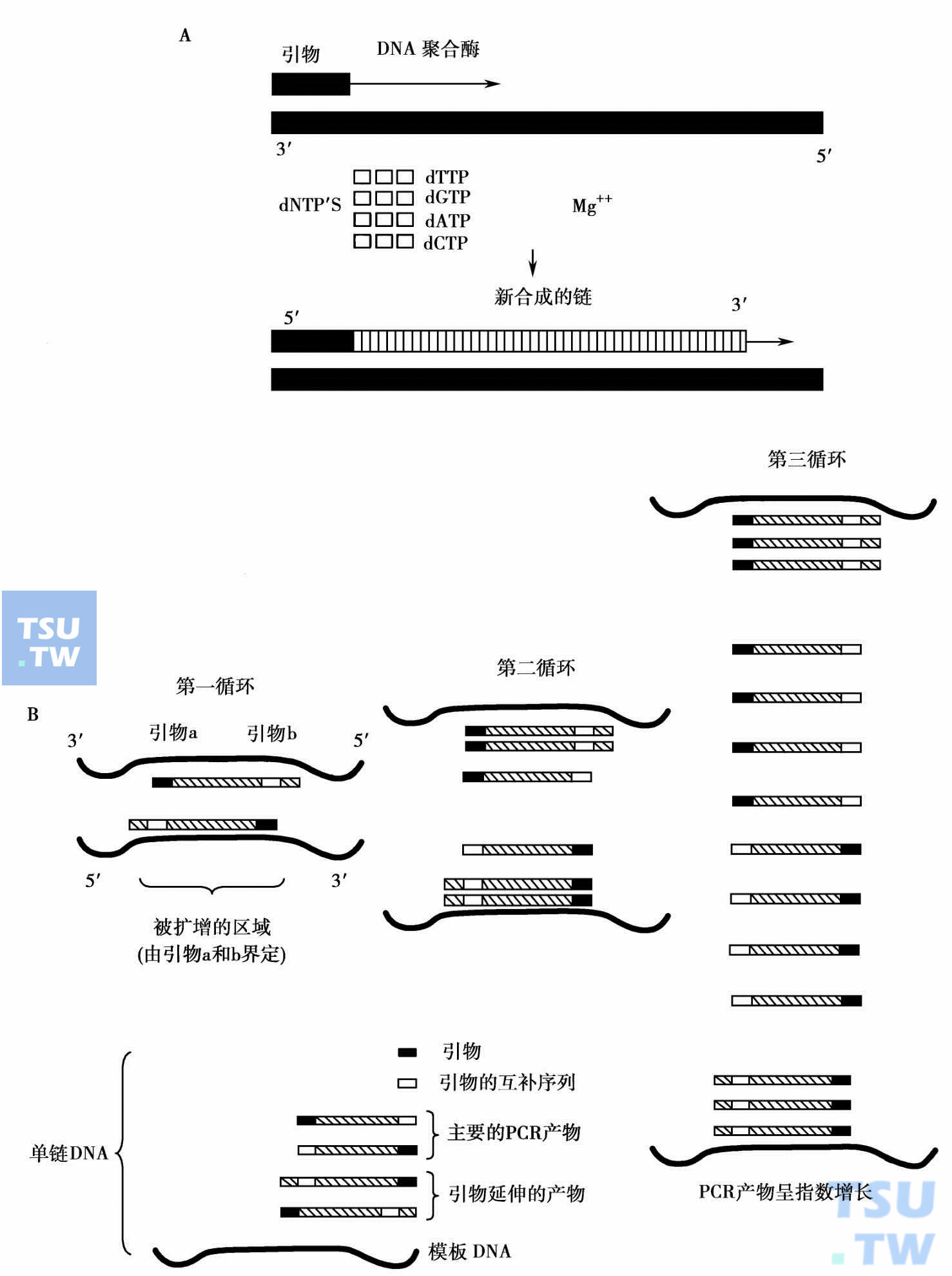
PCR扩增原理:A. DNA聚合酶以互补链为模板延伸引物;B. PCR扩增示意图
PCR自1985年问世以来,由于其所需的样品极少,质量要求比其他基因实验低得多,多种生物技术需要与PCR联用,为了达到不同目的而设计的PCR种类甚多,进展甚快。
基因的克隆分离
从分子水平上研究病因是提高诊断和治疗效果的基础。在基因水平上研究发病机制,首先要有足够量的某一基因DNA。基因工程是20世纪70年代分子生物学中发展迅速的领域。应用基因工程的方法,使致病基因的分离克隆得以实现,从而可以得到大量的某一基因的DNA,大大促进了致病分子机制的研究和对疾病本质的认识。
到目前为止克隆分离的基因已有数千个。主要采取了3种基因克隆的策略:功能克隆、定位克隆和定位候选策略。人们首先克隆的是那些已知生化缺陷或其蛋白质及(或)功能的疾病基因。从分离纯化这一蛋白质着手,测出部分氨基酸序列,然后根据遗传密码推测其可能的mRNA,设计相应的寡核苷酸探针,杂交筛选cDNA或基因组DNA文库,最终获得整个编码区乃至全部基因序列。也可以使用该蛋白质特异的抗体,通过抗原抗体反应筛选克隆于表达载体中的cDNA文库,寻找特异的克隆。上述策略称之为功能克隆。β-珠蛋白基因是第1个被分离克隆的人基因。因为人们对其蛋白功能以及某些生化缺陷已经十分了解,同时在网织红细胞内存在着大量β-珠蛋白mRNA,来源丰富。不少血液病的基因都是应用功能克隆策略获得的。因为如凝血因子FⅧ、FⅨ和vWF等均存在于血液中,它们的蛋白可以从血液中分离纯化。
对于许多疾病,即使是大多数单基因遗传病,基因的生化功能也是不清楚的,因而限制了功能克隆策略的应用。一种新的策略称之为定位克隆的出现,打开了了解疾病的新窗口。仅自1986年至1993年底应用定位克隆策略分离的遗传病基因即已达26个。定位克隆是通过染色体缺失或平衡移位以及DNA连锁分析,确定该基因在染色体上的位置(精确到2000kb左右),然后设法获得覆盖这个基因位点的一组连续的YAC克隆,并在这个区域内寻找基因。获得基因后,从基因的核苷酸序列推出氨基酸序列,回到细胞学和蛋白质化学的研究,进行基因功能的鉴定。由于定位克隆的过程与功能克隆相反,过去曾称为逆向遗传学。
慢性肉芽肿病(CGD)的致病基因就是用定位克隆策略获得的。它是一种中性粒细胞功能缺陷的遗传性疾病。由于缺少活性氧的供应,患者的粒细胞能吞噬细菌而不能杀死细菌。CGD基因的克隆是在完全不清楚其蛋白的情况下,应用连锁分析定位于Xp21. 2-p21. 1,然后克隆了基因。反过来再进行其蛋白的研究和在患者中筛查鉴定。最后,证实了CGD是由于组成细胞色素B的两个蛋白中一个分子量约为91kD的大蛋白缺陷所致(Roger-Pokora B,et al. Nature,1986,322:32-38)。
应用定位克隆策略分离的基因大致属于下列两类:①有大量的家系和用于遗传作图研究的样品;②有明显的缺失或易位,能精确定位的基因。绝大多数疾病不属于以上两类,同时定位克隆要进行大量艰苦的“染色体步移”,工作量很大。而定位克隆目的性又极强,一旦克隆的基因不是预期基因,一切就要从头做起。因此,近年来有人相继提出了新的策略,最受人注目的是定位候选策略。定位候选策略是应用细胞遗传学、医学遗传学和分子遗传学、分子生物学和生物化学的知识特别是人类基因组研究的最新成果,综合了功能克隆、定位克隆和候选基因研究策略,分离鉴定致病基因。下图为定位候选策略的示意图。一旦一个新的致病基因被定位在染色体的某一区域,就可以从计算机的数据库里将在这一染色体区域的所有基因及其有关资料列出一个清单;这些基因可作为该病的候选基因,通过基因特征与疾病特征的比较,可以找到该病最可能的致病基因。然后在患者中筛查该基因是否异常,以证明该基因确为致病基因。同理,当一个新基因定位在染色体的某一区域时,可用上述类似方法进行候选疾病分析。因此这一过程是双向的,可从基因到疾病,也可从疾病到基因。在进行候选时,可以通过OMIM(http://www3.ncbi.nlm.gov/omim/)检索。因此,今后的定位克隆几乎不再需要染色体步移,甚至有可能避开cDNA筛选。
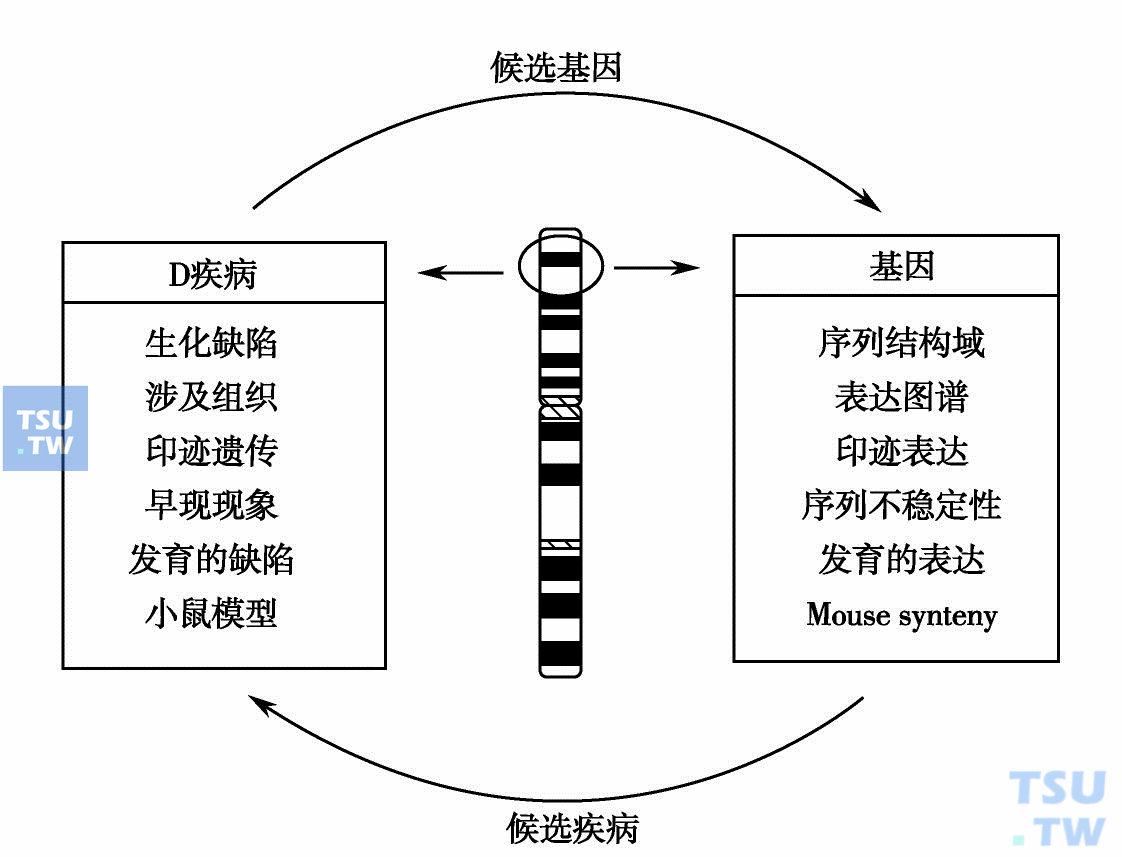
定位候选策略工作程序示意图:当一种疾病被定位于某染色体后在同一区域内分析候选基因,比较候选基因特征与疾病特征
虽然已有数千个基因被克隆分离,但相对人类基因组3万个基因来说,还是很小的一部分,要了解全部致病基因,道路还很艰巨。
