慢性病贫血的发病机制目前并未完全清晰。在慢性疾病过程中,慢性病贫血主要引起机体红细胞生成障碍,不能补偿机体对红细胞的需求。但这种障碍只是轻度的,所以导致的贫血也只是轻到中度。核心的问题是,什么因素导致红细胞生成障碍,铁又是如何被扣留在巨噬细胞和肝细胞中,不能被充分利用。
EPO分泌相对不足及作用钝化
机体针对贫血、组织氧合功能降低的正常反应是代偿性EPO升高及造血增加,一般EPO升高(log)及贫血程度(线性)呈半log相关性。而RA合并慢性病贫血患者的血清EPO水平升高,但是低于IDA患者。血液系统肿瘤及实体瘤合并贫血的研究结果与之类似,提示慢性病贫血患者骨髓反应性代偿不足的一个可能原因就是EPO生成相对不足。支持这一假说的实验有:体外实验发现IL-1、TNF-α通过产生氧自由基而下调转录因子GATA-1(EPO启动子),直接抑制EPO表达。小鼠注射脂多糖(LPS)或IL-1β后肾脏EPO mRNA产生减少、循环EPO水平降低,也证实了细胞因子对EPO生成的抑制。
但并非所有患者都有EPO不足,并且EPO减少并不是AI主要的机制。如果是,则小剂量的EPO治疗即可逆转贫血,然而这并不符合临床治疗的情况,提示体内可能存在红系前体细胞对EPO刺激反应不足。处于慢性炎症状态的肾病血清快速反应蛋白CRP高于20mg/L,所需的EPO剂量较单纯肾病未处于炎症状态的患者升高了80%;另一项研究表示CRP>50mg/L的患者尽管增加了EPO治疗剂量,但贫血仍较CRP<50mg/L的患者的EPO反应性更低,支持炎症导致了EPO的相对抵抗。其他临床研究也发现红系前体细胞对EPO的反应与潜在疾病的严重程度及循环细胞因子水平呈负相关,与前炎症因子抑制红系前体细胞增殖、下调EPO受体及受体后信号传导有关。
红细胞破坏/寿命缩短
一些研究发现慢性疾病患者的红细胞寿命缩短了20%~30%。有学者发现将慢性病贫血患者的红细胞输注到正常人体内,红细胞寿命是正常的,但正常红细胞输注到慢性病贫血患者体内则红细胞寿命缩短,提示是由于细胞外因素导致了红细胞破坏增多。慢性病贫血中大量细胞因子进一步激活了巨噬细胞的吞噬功能以及脾的滤过功能,导致对轻微受损的红细胞破坏增加,这一发现与慢性病贫血外周血中以年轻的红细胞为主也相符合。还有一些其他的因素如细菌毒素、体温升高、宿主来源的抗体或补体介导了红细胞破坏。
铁代谢异常及铁限制性红细胞生成
hepcidin的作用
慢性病贫血发病机制研究中,里程碑式的标志是铁代谢研究的进展。即慢性病贫血患者网状内皮系统(RES)细胞摄取铁增多并引起细胞内铁蓄积,导致循环铁转移入网状内皮系统,继而红系前体细胞可利用的铁减少,引起铁限制性造血。早在1932年即有学者描述感染及低铁血症的相关性,微生物感染时血清铁降低也是一种机体的自我防御机制。向小鼠体内注射白介素1(IL-1)、肿瘤坏死因子-α (TNF-α)或一氧化氮(NO)可引起铁蛋白升高、低铁血症及贫血,提示炎症因子、低铁血症与贫血的相关性,但铁代谢调节参与慢性病贫血的机制一直不清楚。直至2000年,小分子肽段铁调节蛋白hepcidin的发现将炎症因子与铁代谢有机联系起来。hepcidin是肝分泌的抗感染急性相蛋白及铁调节蛋白,小鼠模型中通过转基因或者其他方法诱使hepcidin持续过表达时可导致严重的缺铁性贫血,而缺乏hepcidin的小鼠在矿物油诱发的炎症状态时并未出现血清铁降低,提示hepcidin是慢性病贫血中铁代谢通路的中心环节。进一步研究发现hepcidin通过增加巨噬细胞及肝细胞表面二价金属转运蛋白(divalent metal transporter,DMT-1)以增加铁转运入细胞,同时减少巨噬细胞及肠上皮细胞表面的ferroportin致铁输出减少,从而引起血清铁下降。贫血、缺氧时hepcidin下调,在炎症免疫反应中hepcidin升高。
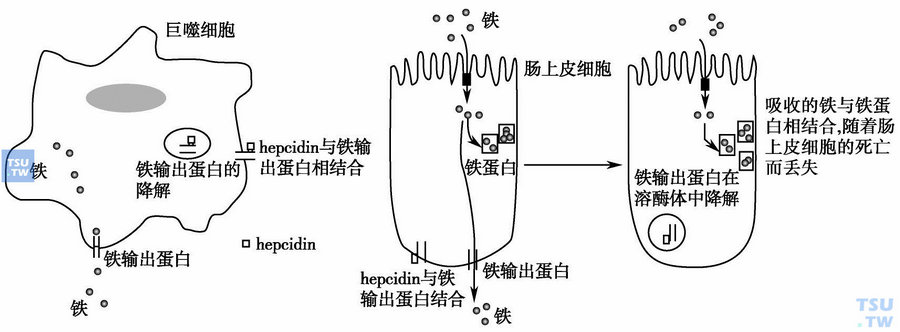
上图:hepcidin作用机制:肠上皮细胞基底面铁输出蛋白ferroportin与铁调蛋白hepcidin结合并内化,导致肠吸收铁降低,血清铁降低;巨噬细胞/肝细胞表面ferroportin减少,铁在网状内皮系统蓄积,血清铁降低。
IL-6、hepcidin及低铁血症
慢性病贫血中多种细胞因子可诱导hepcidin升高,近期研究发现IL-6是影响hepcidin最重要的因素。IL-6基因敲除的小鼠中,在用矿物油处理的炎症过程中未出现hepcidin升高及低铁血症。体外培养的肝细胞中IL-6可有效诱导hepcidin产生,而IL-1或TNF-α并不参与这一反应。在健康受试者中输注IL-6后数小时内诱导hepcidin产生并导致低铁血症。IL-6-hepcidin轴在炎症相关性低铁血症中起重要作用。
血清铁浓度依赖于巨噬细胞及肝细胞的铁释放
稳定状态下机体每日约20~25mg铁进入血浆/转铁蛋白池,几乎均来自巨噬细胞内的衰老红细胞铁再循环以及肝细胞的铁储备,仅1~2mg铁来源于饮食。与转铁蛋白结合的铁仅2~4mg,但是所有铁代谢过程均需通过这个形式,因此转铁蛋白池的铁在数小时内就更替一次。缺乏hepcidin或hepcidin过表达的转基因小鼠中发现hepcidin是铁释放的抑制因子,可同时抑制肠道铁吸收。炎症状态下,IL-6诱导hepcidin生成,随之hepcidin抑制铁释放导致血清铁降低,hepcidin与细胞膜的ferroportin分子结合,并且诱导其内化及降解,后者是铁释放的唯一输出方式。hepcidin浓度越高,ferroportin浓度则越低,肠细胞、巨噬细胞及肝细胞中铁输出就越少。
肠道铁吸收减少
长时间的慢性病贫血患者中红细胞可呈小细胞低色素,部分原因是铁储备逐渐降低导致缺铁、铁限制性造血。肠道铁吸收在炎症状态下被抑制,可能是IL-6及hepcidin介导的。正常人体内储存铁有400~1000mg,每日造血需要的铁仅1~2mg来源于饮食。真正的铁缺乏可能会最终在慢性疾病中出现,尤其在铁储备有限而IL-6水平非常高的儿童患者,例如全身型幼年类风湿关节炎。这部分患者EPO相应升高,但是对口服铁补充治疗无反应,而肠外补铁可纠正部分贫血。
红系前体细胞增殖受损
慢性病贫血患者的红系前体细胞(爆式红系形成单位BFU-E及红系集落形成单位CFU-E)增殖及分化受损,与细胞因子如干扰素-α(IFN-α)、-β、-γ、TNF-α 及IL-1有关。这些细胞因子影响BFU-E及CFU-E的生长,其中IFN-γ是最强的抑制因子,与血红蛋白浓度及网织红细胞数量的负相关性最强。潜在的机制可能涉及细胞因子介导的细胞凋亡,至少部分与神经酰胺的形成有关,后者下调祖细胞表面EPO受体(EPOR)的表达,降低EPO的产生及活性,并减少其他造血细胞因子(如干细胞生长因子SCF)减少。另外细胞因子诱导巨噬细胞样细胞产生不稳定的自由基(如NO)或过氧化物阴离子可对红系祖细胞产生直接毒性。
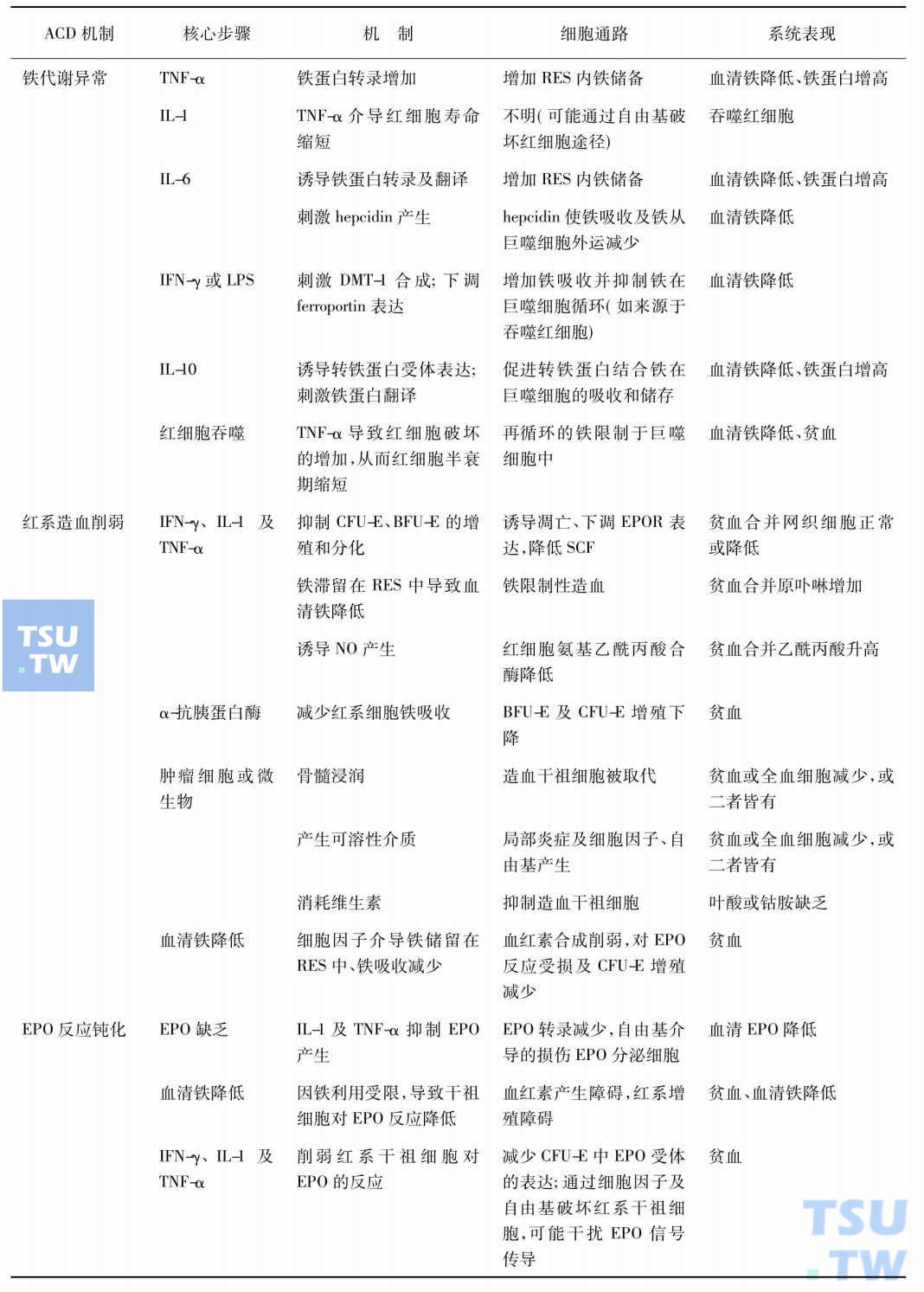
总之,慢性病贫血的发病涉及多个方面,基础疾病可通过一系列细胞因子影响肝铁调节蛋白hepcidin的合成,阻止铁从巨噬细胞和肝细胞的释放,从而造成红细胞生成障碍;红系造血前体细胞的增殖受损;红细胞生成素(EPO)产生减少/反应钝化以及红细胞寿命缩短。各种因素相互影响,最终导致贫血。下表汇总了目前所知的影响慢性病贫血发病机制的因素。
慢性病贫血发病机制及其影响因素