
G-6-PD缺乏症基因突变型
G-6-PD基因位于X染色体(Xq28),核酸长度2269,含有13个外显子,12个内含子,编码区长度1545,共18. 5kb碱基对,编码产物G-6-PD单体由515个氨基酸组成,约59kD,单体无催化活性,形成二聚体和四聚体形式后催化活性。在人红细胞,主要以二聚体存在。
1966年至1986年报告400种以上的G-6-PD生化变异型,分型依据源自异常改变的G-6-PD理化性质、酶代谢动力学、电泳行为等分析指标。1986年至今已鉴定出G-6-PD基因突变型在150种以上,一种基因突变可产生不同的生化变异型,如基因突变型G-6-PD G1160A有6种生化变异型(Iwate、Niigata、Yamaguchi、Beverly Hills、Genova、Worcester),这可能与该酶缺陷在不同人群中表现的多态性有关。G-6-PD基因突变绝大多数发生在编码区即外显子,点突变为主,少数为小片段缺失、终止密码突变、内含子剪接异常等。G-6-PD B是野生型等位基因,一般正常人为B型。
地区性大样本分析各基因型发病率的研究结果表明,G-6-PD基因突变和频率差异很大,基因型有地域性突变热点。在非洲人群,G-6-PD A+(电泳迁移率快于G-6-PD B)是正常黑种人中显现率很高的非洲人种特异性多态等位基因,酶活性基本正常,无溶血;而G-6-PD A-G202A、A376G可出现急性间歇性溶血。非洲裔美国人中G-6-PD A+(A376G)基因频率为11%,G-6-PD A-G202A基因频率为20%。在地中海地区,最常见变异型为G-6-PD Mediterranean (C563T)。在墨西哥,10余种变异型中G-6-PD Santamaria(A376G/A542T)变异型占82%。在西南太平洋地区,G-6-PD Union(C1360T)为常见变异型。在中国人群,突变型有21种以上,基因变异在各民族一致,但发病频率有不同。来自中国内地、中国香港、中国台湾及海外华人的研究数据一致表明,中国人最常见的变异型为G-6-PD Canton(G1376T)、G-6-PD Kaiping(G 1388A)和G-6-PD Gaohe(A95G)。
研究显示,G-6-PD基因突变型绝大多数遗传自非洲、中东和东南亚祖先,均可以追踪到患病家系。G-6-PD新生突变报道很少,仅见近期报告1例新生突变,即G-6-PD Buenos Aires(C 1465T,Pro489Ser,13号外显子),患儿母亲未见相同突变,提示在胚胎发生早期或在母体生殖细胞系发生新生突变。
G-6-PD缺乏症临床表型
最初根据溶血场所和发病急缓将G-6-PD缺乏症分为急性溶血类型和慢性溶血类型,随着对G-6-PD生化变异型和基因突变型的研究,WHO又将急性溶血型分为黑种人高发类型和地中海地区高发类型。1967年WHO根据G-6-PD残余酶活力的高低,将G-6-PD缺乏症分为四型多态性变异型,1989年再次细分为五型。临床为便于诊断,以溶血诱因和临床表现进行临床表型分型。多态性变异型和临床表型的重合与对应关系见下表。
G-6-PD缺乏症分型
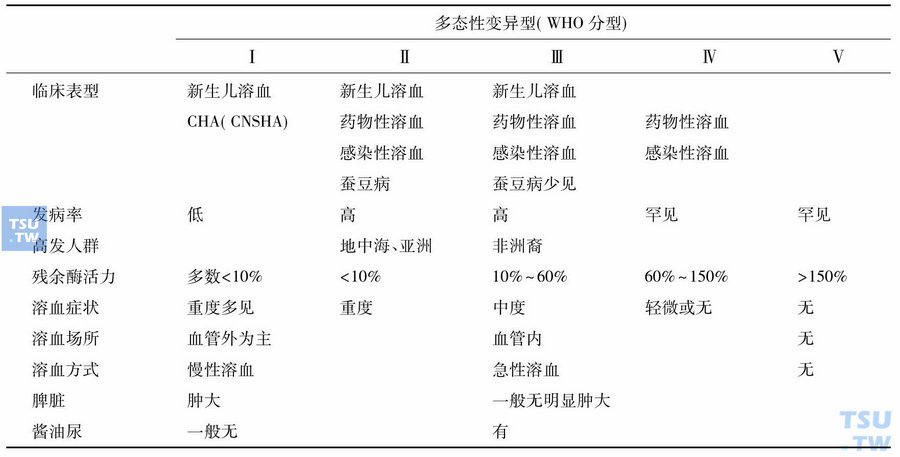
一、G-6-PD缺乏症临床表型分型
根据溶血诱因和临床表现分为五种类型:慢性溶血性贫血(chronic haemolytic anaemia,CHA)、蚕豆病、新生儿溶血、药物性溶血和感染性溶血。其中CHA的临床表现同其他绝大多数红细胞酶病相似,以慢性血管外溶血为主,其余4型均表现为急性血管内溶血。
二、G-6-PD缺乏症多态性变异型分型(WHO分型)
- Ⅰ型:即CHA,表现为慢性溶血性贫血,G-6-PD活力多数低于正常值的10%,溶血严重程度从轻微溶血到依赖输血均见报道,以重度溶血者为多。Ⅰ型在G-6-PD缺乏症中较少见,散在发生。
- Ⅱ型:表现急性溶血,酶活力小于正常值的10%,症状严重。典型代表是G-6-PD Mediterranean,多见于地中海人群如意大利、希腊、西班牙、阿拉伯及犹太(库尔德)后裔,亚洲包括我国等东南亚国家也是高发地区。此型可以引起新生儿溶血、药物性溶血、感染性溶血和蚕豆病。
- Ⅲ型:表现急性溶血,酶活力为正常值的10%~60%,中等程度临床症状。典型代表是G-6-PD A-,非洲地区高发。该类型可以发生新生儿溶血、药物性溶血和感染性溶血,但是很少因食用蚕豆而溶血。
- Ⅳ型:临床症状轻微或无症状,酶活力为正常值的60%~150%,罕见。偶尔可受药物或感染诱导而发生溶血。
- Ⅴ型:有基因突变,无临床症状,酶活力增高,在正常值的150%以上,罕见。
G-6-PD缺乏症基因突变型与临床表型的关联
2000年获得人G-6-PD酶蛋白晶体,对一些突变型进行三维结构分析,利于理解基因型与临床表型的关系、了解G-6-PD缺乏的致病机制。
从酶蛋白高级结构看,每个无催化活性的亚基单体都有底物G6P结合位点和辅酶NADP+结合位点,单体聚合为二聚体和四聚体才具有酶的催化活性。若基因突变影响到酶蛋白底物结合位点、辅酶结合位点或二聚体结合界面的高级结构构象,即可使酶学性质改变而导致临床症状。一般而言,突变距离底物结合位点和辅酶结合位点越近,或突变点保守性越高,患者临床症状越严重。突变后替代氨基酸的电荷性质、极性、pK值等均为影响酶活力的因素。
在G-6-PD酶蛋白结构中,人与其他种属间30%的氨基酸序列同源,其中两个保守基序最具特点:氨基酸残基198~205(RIDHYLGK)为底物结合位点,42~48(GxxGDLx)为构成辅酶结合区域构象所必需的指纹残基。第72位精氨酸(G-6-PDArg72)与辅酶结合直接相关,是保守氨基酸,在23个生物种属保守性高度一致。G-6-PD Pro172保守片段EKPxG为顺式构象,是接近底物和辅酶的关键结构。G-6-PDArg459和Arg463维持NADP+结合区构象、保持酶活性。G-6-PDArg459至碳末端的序列为酶功能所必需。
150余种G-6-PD基因突变造成酶活力缺乏原因大致可分为4种:①mRNA剪接缺陷,直接影响酶蛋白表达量;②酶蛋白折叠缺陷,高级结构构象改变;③不能形成二聚体,而单体无活性;④酶蛋白不稳定。
Ⅰ型变异型即慢性溶血性贫血(CHA)突变几乎都发生在第10和第11外显子,相应于氨基酸序列380~430位,即NADP+结合位点或靠近二聚体界面,影响蛋白折叠,影响疏水性和离子键结合力,酶蛋白表现为热稳定性明显下降,提示NADP+结合对酶稳定性的重要性。
急性间歇性溶血(Ⅱ、Ⅲ型)和较少发生溶血(Ⅴ型)的变异型则散在发生于G-6-PD基因全长序列中,这些突变可影响酶蛋白构象、底物的结合及酶分子的稳定性。如G-6-PD A-型突变影响酶蛋白折叠,与野生型相比,在体外变性后复性时,重新折叠非常缓慢而且不完全,在体内酶的半衰期缩短。G-6-PD Canton(G1376T,Arg459Leu)突变点靠近结构性底物NADP+结合位点,引起严重酶缺陷。
广州中山大学对国内15. 5万人的流行病学调查数据显示,中国人群最常见的G-6-PD Candon (G1376T)中WHO分型Ⅱ型占70%、Ⅲ型占27%、Ⅳ型占3%,G-6-PD Kaiping(G1388A)中Ⅱ型占22%、Ⅲ型占76%、Ⅳ型占2%。
G-6-PD缺乏一般只影响红细胞功能,如G-6-PD A-型红细胞残余酶活力仅有10%,白细胞酶活力却100%。但是有一些突变同时影响有核细胞,均为Ⅰ型CHA,红细胞残余酶活力小于1%时,白细胞酶活力可降至33%以下,例如在C514T (Pro172Ser)突变,可导致白细胞G-6-PD活力仅为正常值的3%,患者G-6-PD严重缺乏、易反复感染。一例患者C269Y突变的等位基因在面颊细胞中也存在,表明出现嵌合现象。这些突变型可能对G-6-PD缺陷与其他疾病和症状相关性研究有提示意义。
