
原发性体液免疫缺陷,即抗体应答缺陷。缺陷由B细胞功能不足,或T细胞不能为B细胞提供适当的信号引起。本组疾病突出的临床表现是反复发生的化脓性感染。同时,对某些病毒易感,可发生中枢神经系统感染和严重的乙型肝炎。
本组疾病主要是先天性B细胞功能不足(发育障碍和功能异常)导致抗体生成缺陷(原发性B细胞免疫缺陷病),典型的例子是X连锁无丙种球蛋白血症。缺陷可以出现在B细胞的不同发育阶段,也可发生在成熟B细胞对抗原刺激应答的各阶段。后者产生的原因是T细胞不能为B细胞提供适当的信号,或者CD8+T细胞功能异常,抑制B细胞产生抗体。此时,B细胞本身多正常。常见的例子是高IgM综合征、普通变异性免疫缺陷病和婴儿暂时性低丙种球蛋白血症。
B细胞功能缺陷的突出和严重并发症是反复化脓性感染。同时,因缺少抗体的中和作用,抗体依赖细胞介导的细胞毒性作用(ADDC)不足,宿主对某些肠道病毒和乙型肝炎病毒的易感,甚至,接种脊髓灰质炎减毒活疫苗可引发疾病。
1. X连锁无丙种球蛋白血症(X-linked agammaglobulinemia,XLA,Bruton病)XLA,由Bruton首次报道,是临床上首次确定的体液免疫缺陷病,也是最常见的原发性体液免疫缺陷病之一。
(1)主要免疫学特征:①发病男性的血液或淋巴组织中只有少数B细胞,甚至无B细胞,扁桃体缺如,淋巴结无生发中心和浆细胞;②出生5~6个月后,母源性抗体下降,开始出现反复化脓性感染;③血清中各类免疫球蛋白缺乏,或只能测得微量的抗体;④细胞免疫正常;⑤免疫球蛋白治疗有很好的反应。
(2)免疫病理及临床特征:多数XLA基因位于X染色体长臂(q21. 3-22)。其编码产物酪氨酸激酶,称为Bruton酪氨酸激酶(BTK)基因,主要表达于B细胞,树突细胞也有表达。该基因由N端类似血小板-白细胞C激酶底物区,一个SH1、一个SH2区和一个C端SH1(酪氨酸激酶)区组成。BTK基因突变方式可以是错义突变、点突变、无义突变和移码缺失(deletion frameshift)。BTK基因缺陷导致B细胞发育过程中传递膜抗原受体传入信号障碍,使骨髓中B细胞的发育停滞在前B细胞阶段前。目前尚未发现影响树突细胞突变和分化的证据。因伴性遗传的X染色体灭活的随机性,有半数B细胞因X染色体携带正常的BTK基因而发育成熟,使女性成为致病基因的携带者。
即使在同一个家族,临床表现也有很大差异。出生6个月后,当母源性抗体达最低水平时,也是患儿逐渐暴露病原体增加的时期,将会出现反复的化脓菌感染,还可出现疫苗相关性多肌炎,单侧膝、踝关节炎和吸收不良性腹泻(与贾第鞭毛虫或轮状病毒感染相关)。尽管T细胞免疫正常,患儿仍对部分肠道病毒和肝炎病毒易感,甚至对脊髓灰质炎活疫苗接种产生病理性反应,发生致死性肠道病毒感染。一种特殊的综合征值得注意,即无丙种球蛋白血症-慢性肠道病毒脑膜脑炎(chronic enteroviral meningoencephalitis of agammaglobulinemia,CEMA),病人有缓慢发展的神经系统症状,共济失调,认知障碍和感觉异常。作为本病的特殊亚型,X连锁低丙种球蛋白血症伴生长素缺乏的基因缺陷位点与XLA一样,是一种连续的遗传性变异。患儿生长激素缺乏,身材矮小。约20%的XLA患者伴有自身免疫病,原因不明。
(3)实验室检查与诊断:血清免疫球蛋白<250mg/dl,其中IgG<200mg/dl,IgA、IgD和IgM难测出。骨髓中可见前B细胞。外周血淋巴细胞缺少SmIg阳性或其他B细胞表面分子,如CD10、CD19、CD20和循环B细胞的EBV受体表达等。缺乏同种血凝素和接种白喉、破伤风、百日咳(DPT)三联疫苗后的抗体应答,锡克反应仍呈阳性。对噬菌体ΦX174的清除和抗体水平都显著低下或缺如。胸腺结构正常,T细胞功能正常。在家系有限的情况下,限制性内切酶片段长度多态性(RFLP)分析,有助于XLA携带者的诊断。外周血循环B细胞的测定可以与婴儿暂时性低丙种球蛋白血症、早期发生的CVID和严重的先天性HIV感染鉴别。
(4)治疗与预后:应用静脉注射免疫球蛋白(IVIG)进行替补治疗。维持血清IgG水平在4g/L以上为佳,至少在2~4g/L。1994年WHO专家组建议的剂量为每次300~400mg/kg,每月1次,或者,150~250mg/kg,每2周1次。除非发生了脊髓灰白质炎、早期不可逆性肺损伤、持续性ECHO病毒感染或淋巴组织恶性肿瘤,早期开始丙种球蛋白替代疗法患者的预后较好。
2.选择性免疫球蛋白同种型缺乏病 抗体产生缺陷只限于一种或少数几种同种型免疫球蛋白的疾病。此类病人对病原体易感性与正常人无显著差别。其原因可以是相应的重链结构基因缺失,或由T细胞缺陷导致。在两个家庭中发现具有κ链缺乏症的患者,其免疫球蛋白只有λ链。
已知的疾病有:选择性IgA缺乏、选择性IgG亚类缺乏和选择性IgM缺陷病。选择性IgM缺陷病是一组病因不同的综合征,临床罕见,发病机制不清。
(1)选择性IgA缺乏(SIgAD):选择性IgA缺乏症是一种因B细胞终末分化障碍引起的疾病,是白种人中最常见的原发性免疫缺陷病,白种人每700人中有1例,北美和欧洲发生率分别为0. 05%~2. 8% 和1:400,在我国,发病率为1:4100,多为散发,有家族史者,多为常染色体显性或隐性遗传。
1)主要免疫学特征:①血清IgA缺乏,分泌型IgA极低;②细胞介导的免疫正常;③免疫复合物病倾向,反复上呼吸道感染,伴消化道疾病和自身免疫病;④其他免疫球蛋白正常或增高;⑤20%患者同时存在IgG亚类缺陷。
2)免疫病理及临床特征:尽管确切病因不清,但具有正常α链基因和表达SmIgA/IgM或IgA/IgD 的B细胞数增加提示,B细胞发育停滞,不能分化成为分泌IgA的浆细胞。分化受阻是B细胞的内在缺陷,还是T细胞辅助功能异常(促IgA分泌的转移生长因子-β)和IL-5产生不足,抑或是B细胞对这些细胞因子的反应低下尚不清楚。群体研究显示,本病与HLA-A1B8BfFC4QOC4BDR3超单元型关联。
本病的临床表现多变。从“健康”IgA缺乏到反复上呼吸道感染,常因程度轻而被忽视。反复肺含铁血黄素沉着症(IPH)也与之相关。另外,由于患者IgA缺乏,黏膜免疫功能低下,多种抗原通过消化道和呼吸道黏膜侵入,易发生过敏性疾病;还与自身免疫病相关,如SLE、类风湿性关节炎、皮肌炎、恶性贫血、干燥综合征和慢性活动性肝炎。本病还有以下现象:①暂时性IgA缺乏见于儿童期成熟延迟,可在5岁后恢复正常;②多数患者即使IgA完全缺乏,其感染机会不增加;③20%有症状的IgA缺乏者,伴IgG2和IgG4缺乏。
3)实验室检查与诊断:血清IgA<50mg/dl,分泌型IgA极低,IgA1和IgA2均减少;其他免疫球蛋白正常或增加。在IgA完全缺乏者,SmIgA阳性的前体细胞数量正常,而IgA1和IgA2仍缺如。这些不成熟表型B细胞,即使在有丝分裂原的刺激下(EBV或pokeweed)也不能成熟为浆细胞。40%病人可有自身抗体。
如果血清IgA缺乏发生在生后第一年,诊断应慎重。因为,完全型共济失调-毛细血管扩张综合征患者的血清IgA缺乏,可早于典型症状4~5年发生。还应与慢性皮肤黏膜念珠菌病、细胞免疫缺陷伴不正常免疫球蛋白合成(Nezelof综合征)和选择性IgG2缺乏相鉴别。获得性IgA缺乏,可见于药物或病毒感染后。
4)治疗与预后:治疗应用由IgA缺乏的无症状患者血中分离的IVIG制剂。
(2)选择性IgG亚类缺乏病:正常情况下,IgG亚类的转换是不同步的,IgG1和IgG3的形成较IgG2和IgG4要快。一旦重链基因缺失或转型异常,将导致一种或多种IgG亚类缺乏。本病病因不清,可能与IgG重链稳定区基因突变、缺失、重组重排障碍或重链基因转录及转录后调节异常有关。血清IgG浓度正常范围较大,但IgG各亚类所占的比例是恒定的:IgG1,60%~65%;IgG2,20%~25%;IgG3,5%~10%;IgG4,3%~6%。WHO的诊断标准是:血清总IgG正常,一种或多种IgG亚类水平异常,低于平均年龄水平的2个标准差。
IgG1缺乏易患化脓菌感染;IgG2缺乏常伴IgG4 和IgA缺乏,儿童多见,患者对多糖抗原反应差,易致反复上呼吸道感染、奈瑟菌属脑膜炎或全身性肺炎链球菌病。值得注意的是,IgG2缺乏时,可出现免疫球蛋白水平正常或增高的自身免疫病。IgG3是最主要的中和病毒的抗体,可单独缺乏或伴IgG1缺乏,成人多见,以反复呼吸道感染为主;正常人群中IgG4值变异很大,即或缺乏,也无症状,因而难以确定选择性IgG4缺乏的存在。由于IgG1占总IgG中的60%以上,所以,当IgG1缺乏时,血清总IgG也减少,常被误诊为普通变异性免疫缺陷病。
诊断IgG亚类缺陷时应注意:①IgG亚类的同种异型变异;②对儿童而言,某些肺炎球菌多糖抗原(6A、9N、14和23F型)只是一种弱抗原,难以产生抗体反应;③对多糖蛋白联合疫苗接种失败的儿童需要检测血清IgG2。
针对IgG亚类患者的感染,一般抗生素治疗即可,不需补充Ig。合并鼻窦炎和哮喘者应以预防发生化脓性鼻窦炎和肺炎为目的,可用IVIG。
3.普通变异性免疫缺陷病[common variable immunodeficiency,CVID,T细胞辅助刺激因子(ICOS),肿瘤坏死因子超家族13B(TNFRSF13B,也称为TACI),肿瘤坏死因子超家族13C(TNFRSF13C,也称为BAFFR)和CD19]作为两性发生率相等异质病,CVID必备的共同特征为抗体产生异常。明显的临床表现常在学龄后期出现。发病率为1:10 000~1:50 000。有家族倾向,遗传方式不明。诊断需要排除任何已知的体液免疫缺陷病因。
CVID基因缺陷、遗传方式和相关的实验表型

BAFFR=B-cell-activating-factor-family receptor
- 主要免疫学特征:①反复的化脓菌感染可发生在任何年龄的人群,明显的临床表现开始于儿童后期;②明显的自身免疫病、淋巴增殖病和消化系统恶性肿瘤倾向;③血清IgG减低,总Ig水平低于300mg/dl。
- CVID的基因缺陷(表62-13):近年,对CVID的基因缺陷研究发现,存在4种基因的缺陷,即可诱导T细胞辅助刺激因子(inducible T-cell costimulator,ICOS)、肿瘤坏死因子受体超家族13B (TNFRSF13B,也称为TACI)和13C(TNFRSF13C,BAFFR)和CD19表达缺失或低表达。
- 免疫病理和临床表现:依据基因表型的差异,临床免疫病理呈现多样性。
ICSO表达于活化的T细胞,缺陷时,几乎没有外周B细胞,类别转换记忆性B细胞缺如,血清呈现低γ球蛋白血症及相关的感染情况。由于缺少ICSO表达的T细胞几乎不能产生IL-10,生发中心的形成缺陷导致记忆性B损伤。ICOS上调CD4和CD8效应器、记忆T细胞和活化的NK细胞,并增强后者的功能。其配体表达于淋巴样和非淋巴样组织(脾T细胞带、淋巴结和Peyer系统)。AR是此型的遗传方式。
TACI/TNFRSF13B缺陷时,淋巴组织增殖是患者的主要临床表型,表现为脾大、扁桃体肥大和IgA缺乏,有15%的患者伴随自身免疫性甲状腺炎。
TACI/TNFRSF13C和BAFF-R配体表达于巨噬细胞、单核细胞和树突细胞。当其缺陷时,将影响NF-κB的激活、免疫球蛋白类别转换的调节和T细胞依赖性抗原的抗体应答。
CD19表达缺陷的患者,其血中B细胞(CD20+)数量正常,而CD19极低表达或缺如,CD27+记忆B细胞和CD5+B细胞减少。缺少对绝大多数抗原的应答性抗体。
CVID与选择性IgA缺乏明确相关。几乎所有CVID患者均有IgA缺乏,一级亲属中常发现有IgA缺乏患者。
儿童期的患者的临床经过与XLA不同,而且其感染的情况也与青春期或成年患者略有不同。多数CVID患者最初只是反复的慢性上呼吸道感染,但反复的下气道感染则应仔细地分析,当发现支气管扩张症时则犹显晚矣(图62-2)。易感病原体与XLA相似,但易感程度低。慢性细菌性结膜炎见于部分患者。合并蓝氏贾第鞭毛虫病明显高于XLA。
不少于50%的患者可出现消化系统的症状,如Crohn病、肠肉芽肿病、肠寄生性细菌或病毒感染、口炎性腹泻和肠淋巴管扩张症等,而严重的吸收不良导致蛋白丢失性水肿可早于CVID的诊断。可有扁桃体、颈部淋巴结和肝脾肿大等非典型淋巴增殖的临床表型。25%的患者易患自身免疫性疾病,甚至出现在诊断CVID之前,如类风湿性关节炎样病症、系统性红斑狼疮、血小板减少性紫癜和溶血性贫血(5%~8%)、皮肌炎、甲状腺功能低下、Graves病、炎性肠病和恶性贫血等自身免疫病,少见的还有白癜风和脉管炎。
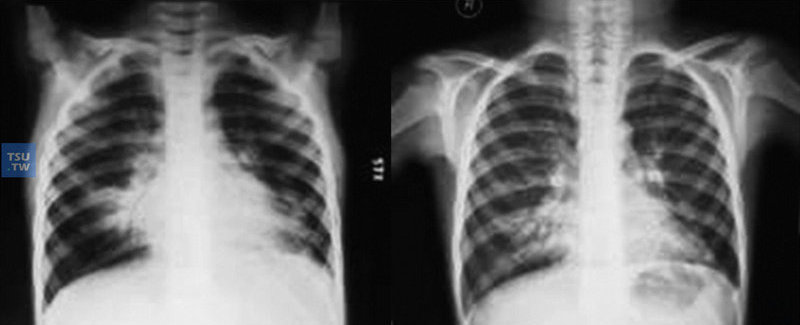
7岁CVID患儿,确诊前反复下呼吸道感染4年。支气管扩张症-CXR-1和2:感染治疗前后胸部X线片比较
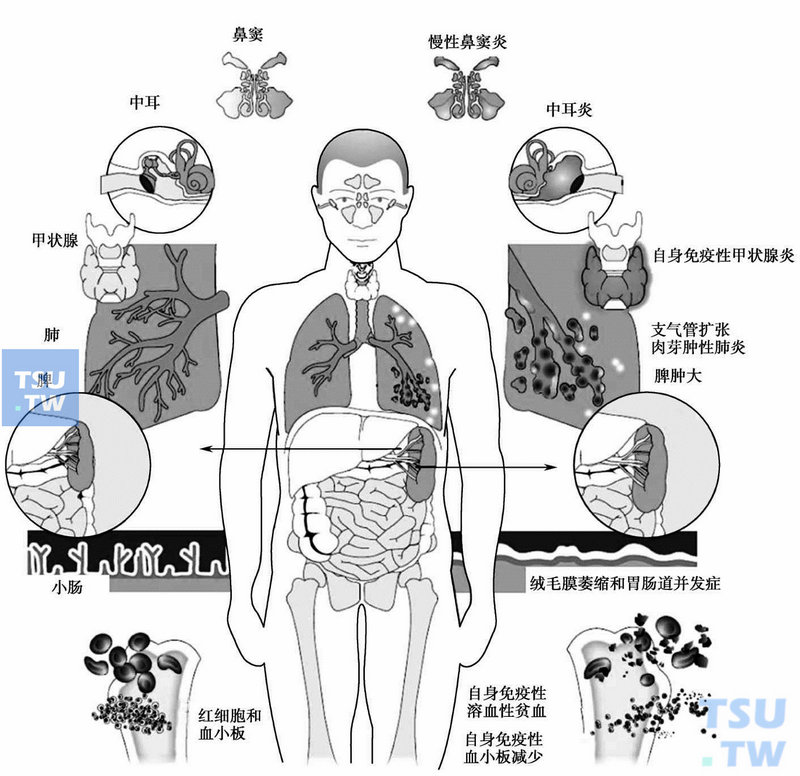
CVID的临床关系
约10%~22%的患者可合并肉芽肿病,并累及多系统。获得诊断的年龄平均在18~34岁。在儿科,大约有三分之一的患儿出现相关的问题。与其他自身免疫性疾病相类似,肉芽肿病也可以出现在CVID获得诊断之前。
CVID患者易患恶性肿瘤。主要是非霍奇金淋巴瘤和消化道肿瘤。
(4)实验室检查与诊断:外周血B细胞数量正常或轻微减少,有时可见缺少T或B细胞表面标志的裸细胞增多。血清IgG<250mg/dl,血清总Ig <300mg/dl。血清IgA减低,IgM通常但并非总是减少。第三种凝集素缺如或降低(<1:10)。特异性疫苗注射后不能产生相应的抗体,锡克试验仍可阳性。因此,减毒活疫苗不能用于CVID的免疫接种。确切的细胞介导免疫异常尚未得到确认,但多数CVID患者的T细胞对抗原应答降低,CD4/CD8比率下降,CD4+CD45RA+(未接触抗原的)T细胞减少。NK细胞活性正常或减低。淋巴组织中缺少浆细胞。部分患者的淋巴结增生,但B细胞生发区的浆细胞缺如与XLA极为相似。
依据诊断步骤的提示,当临床表型和试验检查提示CVID时(第1和2阶段),应对记忆性B细胞和其他外周B细胞亚群的情况进行分析,特别是类别转换记忆性B细胞(CD27+IgMIgD-)低于50%~75%时,支持诊断。
CVID诊断步骤

需要与CVID鉴别的疾病
诊断可以根据上述情况做出,但由于CVID的发病年龄、疾病环节和临床疾病背景的多样性变异,诊断时应除外任何可能影响体液免疫缺陷的病因,包括伴有低丙种球蛋白血症的HIV感染者。当CVID患者以自身免疫病为主要临床表现时,则CVID的诊断和治疗已显延迟。
(5)治疗与预后:CVID患者的治疗与XLA完全相同。治疗的主要目的是减少反复感染的发生率和病死率。以静脉注射免疫球蛋白为替代治疗的选择。通常,每个月一次,300~400mg/kg,使血清IgG水平维持在5. 0~7. 0g/L。合并严重的IgA缺乏时,应考虑选择IgA缺乏的免疫球蛋白制品。对感染的治疗应予以充分的考虑。自身免疫或自身炎症性疾病的治疗则应依照相对应的指南。一般情况下,患者可生存70年以上。女性患者可正常妊娠,生产正常婴儿。主要的并发症是慢性肺病,恶性肿瘤的发生率与是否用IVIG替代治疗无关。
4.婴儿暂时性低丙种球蛋白血症(transient hypogammaglobulinemia of infancy,THI) THI并不是严格意义上的免疫缺陷病,而是发育迟缓逐渐产生正常的过程,婴儿期过后可恢复正常。当来自母体的免疫球蛋白(IgG)以半衰期25~30天的速率逐渐衰减时(3个月时最低),婴儿自身Ig合成逐渐开始。最先产生的是IgM(30周时开始),1岁时达成人水平;5~6岁时,IgG达成人水平,最后是IgA。早产儿在生后3~12个月内,血清IgG可以低至危险水平,而自身合成Ig可延迟到生后36个月。本症婴儿也按上述规律发育,但迟于正常婴儿。主要临床征象是反复发生中耳炎和呼吸道感染,严重的全身感染少见,特殊免疫抗体(包括免疫接种)产生不受影响。本病不需IVIG治疗。
5. Ig重链缺失(Ig heavy chain deletion) 由于先天性重链基因缺失(14q32)导致血清IgG1或IgG2、4缺乏,或伴IgE和IgA缺乏。纯合子患者的血清免疫球蛋白同种型和亚类缺乏,少数患者有反复化脓菌感染;杂合子患者的血清免疫球蛋白亚类轻度缺乏。新近发现,基因的缺失或点突变对重链μ表面限制表达的作用不仅影响IgM产生,还使B细胞分化障碍,导致低丙种球蛋白血症。然而,以此作为新型的免疫缺陷病来解释那些类似XLA的女性患者尚待证明。
6.κ链缺乏(κ chain deficiency) 只有少数病例报告。导致免疫缺陷病的原因只是单纯的κ链缺乏(只有轻链λ的免疫球蛋白)。κ链基因点突变发生在2p11。患者的抗体形成具多变性,除携带κ链的B细胞外,其他循环B细胞数量正常。
7.免疫缺陷伴胸腺瘤(immunodeficiency with thymoma,Good’s syndrome)主要免疫学特征是反复感染伴随先于胸腺瘤发生的获得性低丙种球蛋白血症。以反复的上呼吸道感染、慢性腹泻、皮炎、败血症、胃炎和尿道感染为主要的临床表现。伴胸腺瘤的患者常合并肌无力(重症肌无力)、再生障碍性贫血(也可见纯红细胞发育障碍性贫血)、血小板减少、粒细胞减少、自身抗体形成、糖尿病、淀粉样变性、慢性肝炎和非胸腺癌肿。早期,低丙种球蛋白血症呈间歇性发生,多在常规胸部放射检查中偶然发现胸腺肿瘤。75%的胸腺瘤细胞为纺锤形,部分呈恶性。血清免疫球蛋白严重缺乏,抗体应答低下,部分患者外周血淋巴细胞对PHA反应缺如、迟发皮肤过敏试验无反应。可出现抑制性T细胞活性增强。切除胸腺瘤并不能纠正免疫缺陷,但纯红细胞发育异常和重症肌无力则会得到改善。IVIG可用于反复感染和腹泻的治疗。
8. 5’-核苷酸酶缺乏(5’-nucleotidase deficiency)研究发现,在获得性或XLA、Wiskott-Aldrich综合征、AIDS和SIgAD的部分患者中,其5’-核苷酸酶活性减低。后者,作为外周血淋巴细胞,特别是B细胞的分化标志,活性的减低反映了患者B细胞数量的减少和突变的某种异常。
9.转钴胺Ⅱ缺乏(transcobalaminⅡdeficiency)转钴胺Ⅱ,一种维生素B12结合蛋白,是维生素B12进入细胞所必需的。一旦缺乏转钴胺Ⅱ,病人将出现低丙种球蛋白血症、巨幼细胞性贫血、淋巴细胞减少、粒细胞减少、血小板减少和严重的肠吸收障碍。
