
又名蕈样霉菌病(mycosis fungoides,MF),是起源于记忆性辅助性T细胞的低度恶性皮肤T细胞淋巴瘤,约占所有原发性皮肤T细胞淋巴瘤的50%,其组织病理学特征为具有脑回状胞核的中小淋巴细胞的亲表皮性浸润。本病少见,国外报道本病年发病率为0.3/10万,男女发病率之比为(1.6~2.0)∶1。多累及老年人,确诊时平均年龄55~60岁,但儿童及青年人也可发生。
本病首先由1806年法国Alibert报道,因部分皮肤肿瘤形似蘑菇,当时误称为蕈样雅司,1835年更名为蕈样霉菌病。1876年Bazin描述了本病的典型临床表现,即Alibert-Bazin型,其皮损有三期表现,开始为斑片,以后发展为浸润性斑块,最后可出现肿瘤、破溃,其自然病程可达二三十年以上。1885年Auspitz提倡用蕈样肉芽肿,沿用至今。同年Vidal及Brocq曾提出另一型,即所谓暴发型,现已明确该型为其他类型皮肤T细胞淋巴瘤或皮肤B细胞淋巴瘤。1892年Hallopeau又提出第三型,即红皮病型,目前认为该型多为Sézary综合征。
病因及发病机制
本病病因及发病机制尚未阐明,研究提示遗传、环境、免疫因素等参与了本病的发生。
- 遗传因素 逐步累积的遗传异常导致淋巴细胞出现克隆增殖、恶性转化,最后形成淋巴瘤。目前已发现多种遗传因素参与了本病的发生。染色体结构异常(如:易位、缺失)可引起癌基因的激活和抑癌基因的失活,这是肿瘤发生的重要分子基础。研究表明t(12;18)(q21;q21.2)染色体易位可导致NAV3基因断裂,半数以上患者的皮损中存在该基因的缺失,晚期患者缺失率更可高达85%,据推测该基因可能为抑癌基因,其缺失可能与MF肿瘤细胞的异常增殖、分化有关。应用比较基因组杂交方法发现,MF患者可存在1p,10q,17p,19,13q染色体缺失,或17q,4染色体获得,其中10q23.33-24.1和10q24.33-25.1两个区域的缺失频率高,提示该区域存在与本病发生密切相关的基因。另外,肿瘤细胞中某些基因(如DNA错配修复基因MLH1)启动子的高甲基化所导致的基因表达障碍也参与了本病的发生。
- 环境因素 持续的抗原刺激参与多种恶性淋巴瘤的发生,如EB病毒与鼻NK/T细胞淋巴瘤,乳糜泻与肠病型T细胞淋巴瘤,伯氏疏螺旋体(Borrelia burgdorferi)与皮肤B细胞淋巴瘤发生关系密切。特应性皮炎患者似乎更易患蕈样肉芽肿,提示持续的抗原刺激同样可能也是本病的启动因素,但其抗原尚未确定。既往曾认为HTLV-1、EB病毒参与了本病的发生,但多项研究并不支持该假说。然而,有研究提示细菌感染参与了本病的发生,该研究发现,在以红皮病为主要表现的MF/Sézary综合征患者中,76%可自血液或皮肤中分离出金黄色葡萄球菌,其中半数细菌携带编码内毒素(如:中毒性休克毒素-1,即TSST-1)的基因。另外,有研究表明某些化学致癌剂(如石油产品)的职业暴露与本病发生相关,但也有研究不支持该论点。
- 免疫因素 大多数MF病例的肿瘤细胞起源于记忆性辅助性T淋巴细胞,MF细胞通过与真皮血管内皮细胞相互作用归巢至皮肤。其基本过程如下:因MF皮损中的血管内皮细胞处于活化状态,其表面表达E-选择素,而血液循环中的MF细胞表面表达其配体——皮肤淋巴细胞抗原(CLA),从而介导MF细胞在血管内皮细胞表面滚动。然后,在基底细胞分泌的趋化因子CCL17作用下,MF细胞依靠表面的趋化因子受体CCR4游走出血管并进入表皮。进入表皮的MF细胞表现出与表皮细胞的高亲和力,并可簇集在朗格汉斯细胞周围而形成特征性的组织病理表现——Pautrier微脓肿,该过程由MF细胞表面的整合素aEβ7、趋化因子受体CCR4、CD4 TCR复合体分别与上皮钙黏蛋白(E-cadherin)、趋化因子CCL22、MHC Ⅱ类分子结合介导。
研究表明MF细胞表达CD45RO、PCNA、CD25等活化标志,处于持续活化状态,并分泌多种细胞因子。早期皮损中的细胞因子以Th1型为主,肿瘤细胞中含有较多CD8+T细胞,它们通过细胞毒作用,或分泌IFN-γ等细胞因子在抗肿瘤应答中具有重要作用。事实上,真皮浸润细胞中具有高比例CD8+T细胞者预后较好。这些细胞表面表达FasL,可与MF细胞表面的相应受体Fas结合而介导其凋亡,但不表达Fas的MF细胞则可逃避免疫系统的攻击,这也是MF众多免疫逃逸机制之一。与早期不同的是,肿瘤期皮损中的细胞因子以Th2型为主,这一微环境抑制了抗肿瘤的细胞免疫应答。
临床症状
典型蕈样肉芽肿的病程呈慢性进行性,自红斑期进入斑块期,最终发展至肿瘤期可达数年甚至数十年之久。
红斑期
又名蕈样前期。其皮损早期表现可分为两种类型:
非萎缩性斑片
扁平、淡红色、鳞屑性斑片,直径数厘米。类似银屑病或某些类型皮炎。此型进展较快,可数月或数年进入斑块期,甚至出现内脏病变。
蕈样肉芽肿,斑片期色素性皮损
萎缩性斑片
表面萎缩、光亮或出现皱纹,伴有毛细血管扩张、色素增多或减少,可类似斑片状副银屑病的大斑片或斑驳状副银屑病。此类型皮损可长期存在,无大变化。据统计,仅12%病人可进一步发展,在斑片基础上出现不规则浸润,从而进入斑块期。
以上两型有时不能截然区分。皮损较明显时,皮疹则往往多形,可以在一病人身上同时存在,如红斑、丘疹、苔藓化、鱼鳞病样或皮肤异色样损害。少数病人尚可见风团、水疱或紫癜样皮疹。因此,临床上可类似银屑病、副银屑病、湿疹、脂溢性皮炎、神经性皮炎、鱼鳞病或皮肤异色症。个别病人尚可类似玫瑰糠疹、麻风、肥大细胞增生症、丹毒或红斑狼疮。然而最常见者为红斑鳞屑性损害或萎缩性斑片,边缘清楚,但不规则。早期损害可群集,中央可消退而向四周扩大,或排列成弧形、环状、半环状,也有如地图状或带状者。皮疹可分为红色、黄红、淡褐色,多伴有色素沉着或减退。皮损最好发于躯干,但其他各处均可发生,也有长期限于大腿内侧者。
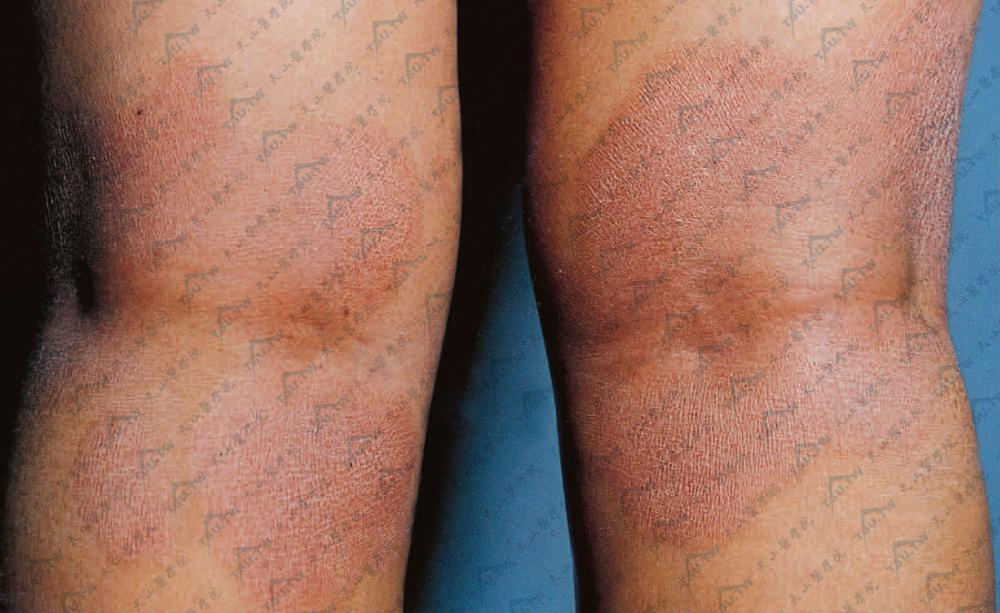
又称蕈样肉芽肿前期。皮损多形。腘窝处境界清楚的红斑,有鳞屑,似湿疹、皮炎改变,有轻度浸润,瘙痒剧烈
瘙痒常为早期或惟一的自觉症状。这种瘙痒常难以忍受,常规治疗难以缓解,并且可持续存在,甚至可长达10年,故严重瘙痒对本病的早期诊断有一定帮助。但也有不少患者不痒或不经常痒。此期通常持续2~5年,少数病例可以非常短暂,但也有长达30年者。
斑块期
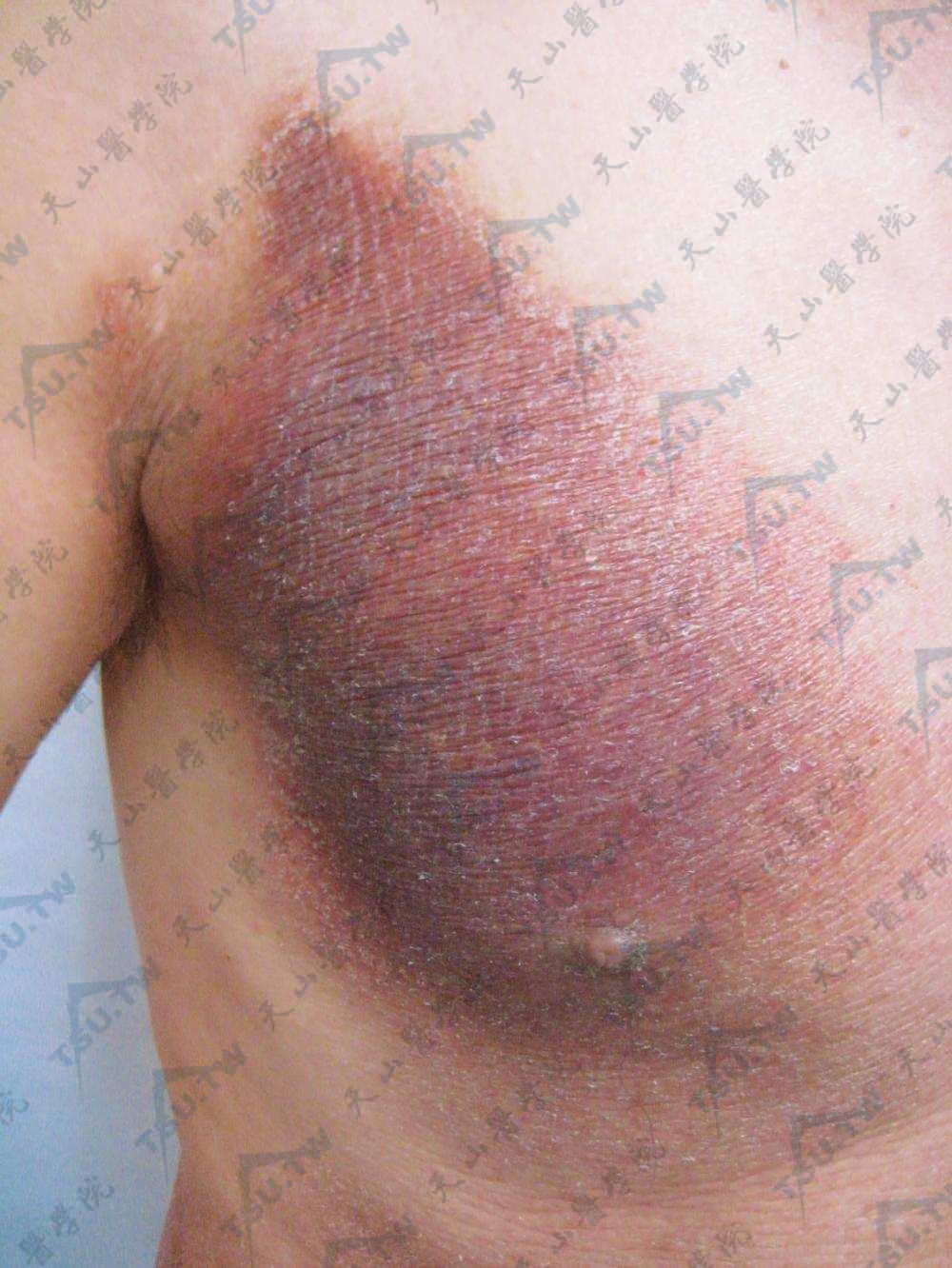
又名浸润期。本病可因第一期极其短暂而不明显,一开始即为第二期,但通常皆经第一期后再进入第二期。此期浸润不断增加,往往呈暗红厚垫状、不规则形隆起斑块,表面紧张、光亮、高低不平,甚至也有呈疣状或表面反复渗出结痂而呈蛎壳状。浸润斑块可泛发全身,也可局限于某些原有皮损部位,或伴有丘疹或小结节。皮疹颜色也各不相同,可为淡红、黄红、砖红、紫红、暗红乃至棕红或褐色。浸润斑块可以不发生破溃,或数日内破溃,也可在出现新的浸润斑块时发生破溃。陈旧浸润斑块偶可自行消退不留痕迹。浸润斑块表面毛发常见脱落。此外,值得注意的是,不同斑块,甚至同一斑块的不同部位,其浸润程度往往不同。例如环状或半环状损害可以一部分与正常皮面相平,而另一部分因显著浸润而隆起。有时在红斑基础上出现不规则的浸润或散在小结节状浸润。这些特殊表现对诊断有意义。在此期内通常明显瘙痒,除少数浸润可自行消退并留下萎缩及色素沉着或减退外,一般浸润损害常持续存在,甚至增生如疣状。
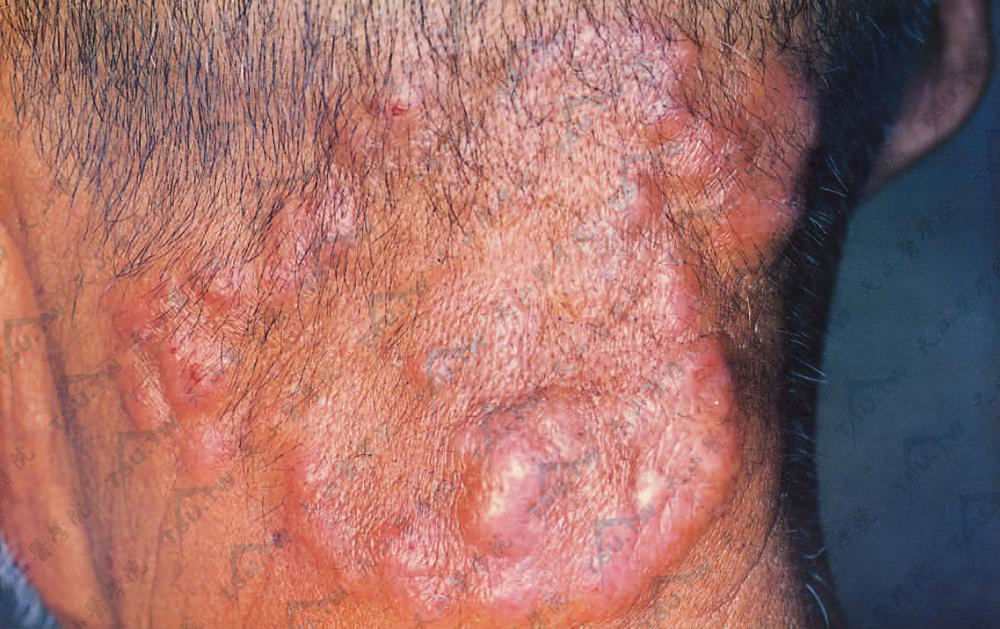
颈项部浸润性结节,融合成大片斑块
肿瘤期
通常在浸润损害的基础上逐渐出现肿瘤,常常在陈旧浸润损害的边缘或中央发生,很少在新起的浸润损害上出现肿瘤。肿瘤可向表面隆起,甚至如蕈样,时有破溃,也可以如半球状,其基底部浸润范围较宽广。肿瘤可迅速增大,数目增多。直径大小不一,大者可达数厘米。颜色可为灰白、黄红乃至棕红色。肿瘤多见于面、背及四肢近端。完整的肿瘤一般无痛感,但破溃者常有剧痛,破溃后可留下萎缩性瘢痕,伴有色素改变。
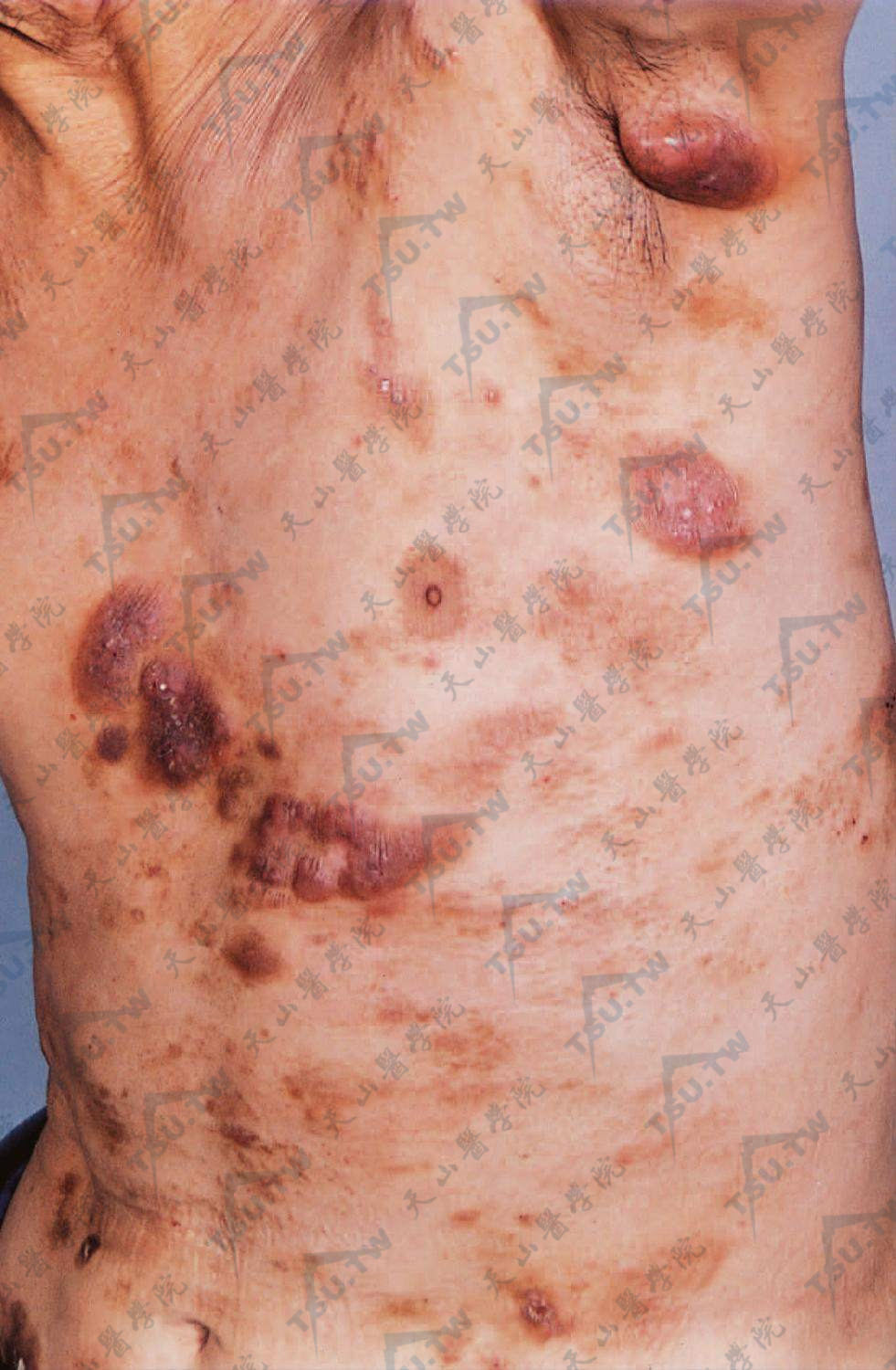
在浸润斑块基础上出现大小不等的肿瘤,可达鸡蛋大,同时有多数浸润性斑或斑块,暗红色
此外,本病除皮损区域内毛发可以脱落外,可出现全秃,或不同程度的毛发脱落。也可出现毛囊性黏蛋白变性,表现为正常肤色或暗红色由毛囊性损害融合组成的斑块,多见于头皮、面、颈,伴有明显毛发脱落。黏膜很少侵犯。极少数病例发生口腔或齿龈黏膜损害。MF患者可见淋巴结肿大,早期为皮病性淋巴结病,晚期淋巴结可受累。此时往往内脏器官也同时有病变,几乎所有内脏器官均可被侵犯,例如肝、脾、肺、肾、骨髓等,其他脏器病变也有报道。末梢血常规一般无特殊变化。晚期病人可见贫血。个别报道有嗜酸性粒细胞、淋巴细胞或单核细胞增高者,后者往往是晚期患者的表现。通过免疫学的观察发现各类淋巴瘤患者的淋巴细胞转化均受到抑制,但以本病最轻,早期可无异常,病情进展时反应低下,似与皮疹广泛及病期有关。而体液免疫方面,一般只有在晚期才受到损伤。
本病病程呈慢性进行性,个体差异大,有的时轻时重,或缓解与加重交替,可长达数年乃至二三十年,甚至个别达30年以上者。多数因恶病质或并发严重感染或化疗反应而死亡。
组织病理
组织病理变化也可分为3期:
红斑期
和临床表现一样,病理变化也多种多样。萎缩与不萎缩的斑片有所不同。虽然有人认为MF一开始就是淋巴瘤,而不是从其他皮肤病发展而来,但早期MF肯定诊断是困难的,特别是扁平不萎缩的斑片,有时往往需要多次取材作病理检查,才能确诊。
扁平、不萎缩的斑片中,开始在真皮乳头及乳头下层仅见单纯性炎症浸润。浸润主要是淋巴细胞,但也含多少不等的组织细胞。但早期病变中,时常可见亲表皮现象(epidermo tropism),此现象高度提示为MF病变。出现亲表皮现象时,可见表皮内散在单个的单一核细胞,与周围的角质形成细胞之间有一透明晕,将其分开,偶尔可见几个单一核细胞密集在一起,周围有晕,这提示为小的Pautrier微脓肿。通常看不到核深染而形态不规则的MF细胞,即使浸润中有少数这样的细胞,也不能肯定MF诊断,因为在各种炎症皮肤病(如扁平苔藓等)的真皮浸润中偶尔也能出现此种细胞,即所谓脑回状单一核细胞(CMC)。
在萎缩性斑片中,其组织相类似血管萎缩性皮肤异色症,可见表皮变平,基底细胞空泡化,紧接表皮下有带状单一核细胞浸润,在有些区域侵入表皮。此外,可见毛细血管扩张、红细胞外溢与色素失禁。MF早期皮肤异色症样皮疹的浸润可以无特异性,但浸润较伴发于皮肌炎或红斑狼疮的血管萎缩性皮肤异色症皮损者为著。在比较晚一些的皮损中,在真皮浸润及表皮内可见深染、核扭曲的细胞。在表皮内大多为单个有晕的细胞,也可见细胞聚集,但通常仅偶可见明显的亲表皮现象。
斑块期
在大多数病例中,此期的组织相有诊断价值。通常可出现下列3种变化:
- 亲表皮现象,单个散在深染脑回状单一核细胞,周围有晕,往往在表皮内出现Pautrier微脓肿;
- 真皮浸润呈带状或斑片状;
- 真皮浸润内出现相当多的所谓MF细胞,即其核深染,外形、大小不规则,因此呈异形表现。
非常有帮助的诊断特征是表皮中出现亲表皮现象及Pautrier微脓肿,特别是多次活检,大多数斑块期病变中均可见到。亲表皮现象的特点是表皮内有散在单一核细胞,其周围往往有一晕状透明间隙。Pautrier微脓肿是由紧密聚集的单一核细胞在表皮内形成小群,周围也有透明间隙,因此甚似成群细胞位于空泡之中,这与红斑期浸润中亲表皮现象稍有不同,红斑期中进入表皮的细胞近似普通的淋巴细胞,而斑块期进入表皮的细胞,有些已有MF细胞的表现。亲表皮现象不仅见于表皮,有时在毛囊上皮中也可见到。
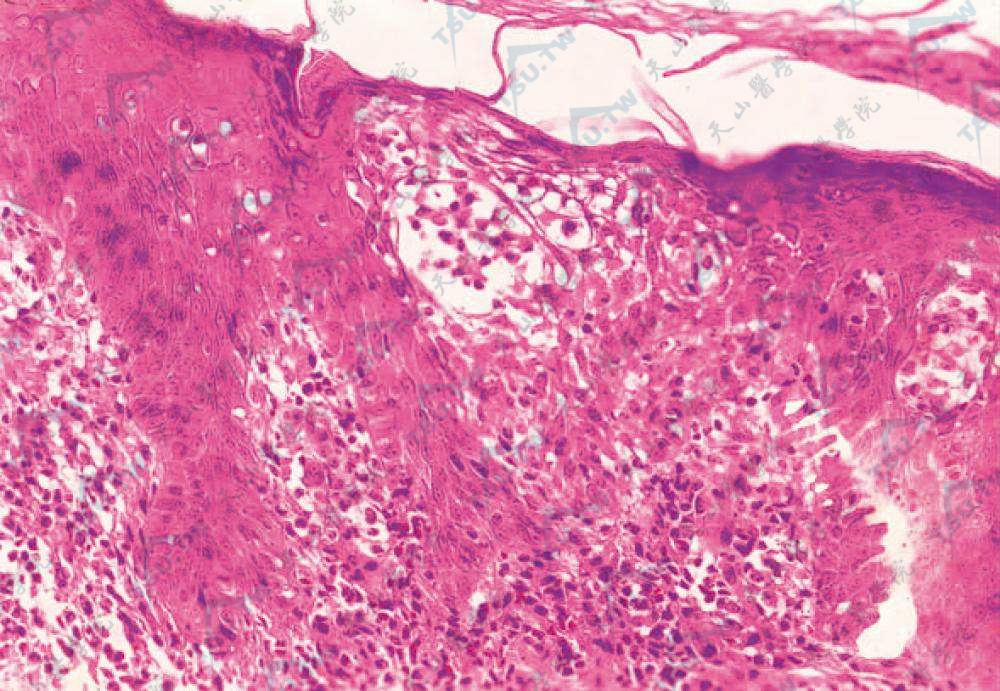
肿瘤细胞侵入表皮内,形成Pautrier微脓肿(HE染色×400)
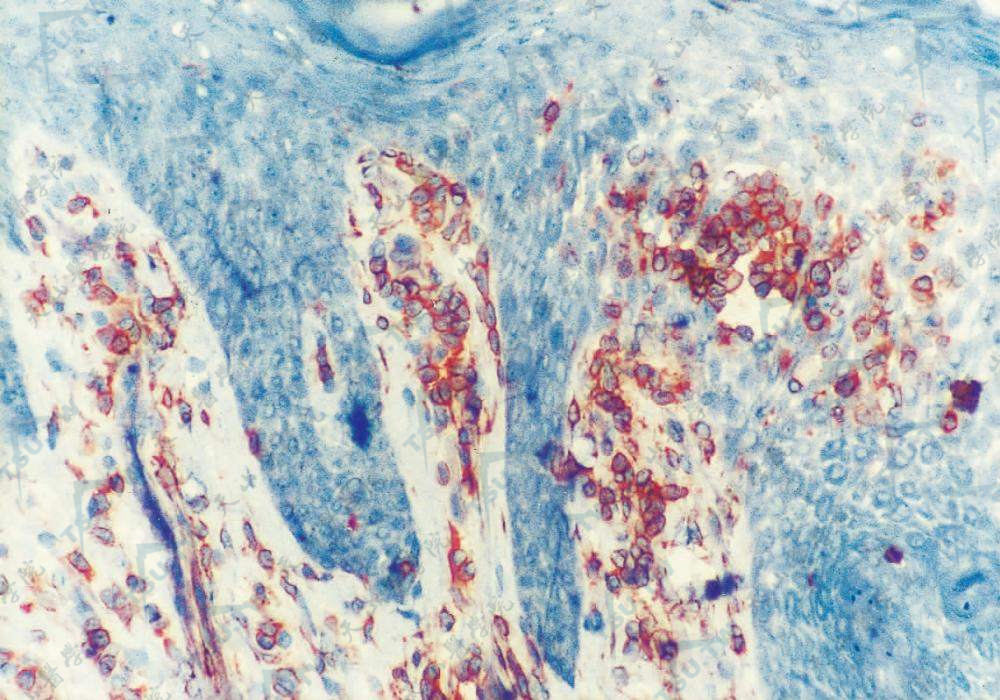
免疫组化见多数CD4+细胞(×400)
斑块期中,细胞浸润的排列呈带状或斑片状,如斑片状相当大而且境界很不清楚,这是倾向于诊断MF的有力依据。浸润细胞成分,除淋巴细胞外,还可见到组织细胞,多少不等的MF细胞,有时上述细胞不易辨认,故统称单一核细胞,有些病例尚混有嗜酸性粒细胞及浆细胞。
出现MF细胞对斑块期MF有诊断价值,特别是真皮浸润中具有相当比例时更有意义。有些MF细胞的核并不显著大于斑块期浸润中其他细胞的核,因此MF细胞之所以突出,实际上更凭借其核深染与扭曲,而大小在其次。但在光学显微镜下大者容易识别,小者常不易肯定,往往在电子显微镜下才能证实,并偶可找到MF细胞的核有丝分裂象,但为数不多。只有到斑块晚期,MF细胞的核才较浸润中其他细胞核明显为大,此为典型的MF细胞。
MF斑块期病变中,有时在毛囊及皮脂腺的上皮细胞出现变性,其中可见酸性黏多糖,形成黏蛋白秃发或毛囊性黏蛋白病,此种黏蛋白变性可能是单一核细胞侵犯毛囊皮脂腺的继发结果。
肿瘤期
亲表皮现象不明显,真皮内有大片浸润,往往深达皮下组织。浸润可压迫并破坏表皮,使其形成溃疡。大多数病例中,浸润主要由MF细胞组成,核异形,深染,大小有显著差别。然而有些病例许多细胞出现母细胞转化,具有大而呈泡状的核及明显核仁,因此类似组织细胞。这些细胞常见核有丝分裂象。偶尔,母细胞也有异形,并表现为单核或多核巨细胞,可类似霍奇金病中的核仁。此时诊断MF则较困难,因肿瘤期往往没有亲表皮现象。
诊断及鉴别
主要根据临床上的特点与组织学的指征,早期诊断一般均需作活检确定。因此当临床上怀疑为本病时,应及时作活检,且往往需连续切片方能找到特异性病变。Farber曾提出用“削片法”连续切片观察表皮病变。也有学者建议早期浅表病变可用锐利的刀片仅削取病变处的表皮,连续切片寻找Pautrier微脓肿,同时作细胞涂片检查。此法比较简单,对患者损伤较少,不需要缝合,有利于反复取材。但即使如此,有时仍不易确诊,故早期诊断方法仍有待于进一步研究。因此对本病诊断应慎重,应临床与病理及免疫组化结果密切结合,必要时密切随访,多次取材,切不可主观片面,草率从事。目前,下列诊断指征可供参考:
临床方面
- 皮疹多形,而且同时存在,以致临床上往往既像某一病,又像另一病;或者既不完全符合某一病,也不完全符合另一病。换言之,这些皮疹表现很难用一种皮肤病来解释或概括。例如在一患者身上有些损害类似皮肤异色症,而另一些损害又像鱼鳞病。如为红斑损害,则常为暗红色或棕红色。既增生肥厚,又伴有萎缩。而且常有色素异常、色素沉着与色素减退并存。皮疹边缘不规则,形态也不一致,分布又无规律。这些都是本病的特点。
- 虽然瘙痒在本病开始或病程中并非必有的症状,但不少病例均有瘙痒。特别是皮疹泛发而有顽固性剧痒,用一般药物难以控制者,应怀疑有无本病的可能性。
- 蕈样肉芽肿虽然部分皮疹可以自然消退,但总的来看,皮疹不断增多,浸润逐渐加重,因此呈慢性进行性的过程,病程往往较长,也是其特点之一。
病理方面
- 亲表皮现象。真皮内浸润细胞往往侵入表皮甚至毛囊上皮,即所谓亲表皮现象,侵入表皮的单一核细胞,周围有晕,并有聚集形成Pautrier微脓肿的趋势。这种现象为本病诊断的重要依据之一。这种亲表皮现象与一般湿疹、皮炎所见的细胞外移不同,后者往往伴有海绵水肿,细胞多为中性粒细胞、淋巴细胞混杂在一起,而MF则无明显水肿,全部为单一核细胞。
- MF细胞斑块期开始真皮内即可出现相当比例的MF细胞,核深染、形态不规则,甚至大小不一,细胞周围有透明晕,对诊断有价值。
- 浸润形态呈T细胞模式。早期浸润多限于真皮上部,多呈带状,伴有基底细胞液化,类似皮肤异色病的表现者,往往要考虑本病。
在鉴别诊断方面,因本病可类似很多皮肤病,故不能一一叙述,可结合临床与病理所见分别加以排除,必要时需要观察病程,不同时期多次取材,才能加以鉴别。有学者提出早期MF的组织病理学诊断标准(下表),但多数病例需结合临床方能确诊。
早期蕈样肉芽肿的组织病理学诊断标准

另外,在大多数病例中,应用Southern印迹或PCR可检测出克隆性T细胞受体(TCR)基因重排,对诊断也有帮助。一般而言,TCR基因重排几乎能够100%地预测肿瘤期、50%~100%斑块期以及50%~78%斑片期的蕈样肉芽肿。但对该结果的解释应该谨慎,因为TCR基因重排在一系列炎症性皮肤病时也有发生,包括扁平苔藓和急性痘疮样苔藓样糠疹。
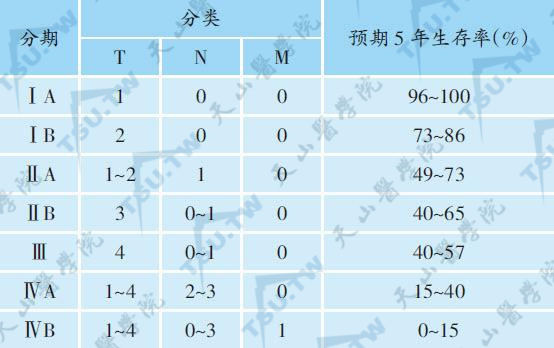
蕈样肉芽肿除临床及病理上分为3期外,为了进一步观察病情,选择适当的治疗和评定预后,先后提出了进一步分期的方法。1975年美国MF研究协作组提出TNM分类方法,此分类可以反映出预后意义,同时也比较合理,可以重复,现介绍如下(下表)。
CTCL的TNM分期标准

CTCL的分期分类与预后
为准确分型,需完成以下工作,包括:完整的病史,全面的体格检查(特别应注意认真进行淋巴结触诊),全血细胞计数及异型淋巴细胞(Sézary细胞)计数,血清生化检查(包括肝肾功能,乳酸脱氢酶),胸部放射线检查,皮肤活检。如可触及淋巴结肿大应进行组织学检查,但为避免假阴性不推荐应用细针穿刺活检。如上述检查发现异常,应进一步检查。CT扫描可用于评价Ⅱ~Ⅳ期患者胸部、腹部、盆腔、内脏有无受累,但IA期患者无需进行该检查。
本病病程呈慢性进展性,多数患者预后较好,各期预后不同,其预期5年生存率见上表。淋巴结肿大、肿瘤、皮肤溃疡是主要的预后评价指标,如无其中任一表现则不会直接死于该病;而一旦三者全部出现,其中位生存期仅1年。
治疗
治疗目的包括清除皮损,以提高生活质量、延长无病生存率及总体生存率。早期可采用仅对症处理的“期待疗法”,或皮肤靶向治疗(skin-directed therapy);一般仅晚期患者方考虑化疗。具体方案可参考EORTC 2006年制订的蕈样肉芽肿推荐治疗方案(下表),根据临床表现和治疗手段的可用性选择具体的治疗方法。
蕈样肉芽肿的推荐治疗方案(摘自EORTC,2006)

注:治疗方案的推荐顺序基于该方案全体作者的共识。具体治疗方法根据临床表现和治疗手段的可用性选择。
皮肤靶向治疗
- 外用糖皮质激素 糖皮质激素,特别是Ⅰ类(超强效),用于红斑期MF疗效好。其作用机制主要包括诱导肿瘤T细胞凋亡,以降低肿瘤负荷;并可通过减少朗格汉斯细胞数量,从而减少对肿瘤T细胞的刺激。
- 外用氮芥 对红斑期和斑块期皮损疗效好,对肿瘤皮损也有一定疗效。通常应用0.01%或0.02%氮芥溶液或软膏。氮芥溶液常用生理盐水或蒸馏水稀释(10mg/50mL)后外用,每日1次,药液最好新鲜配制,放冰箱中保存,皮损消退后,仍需要维持治疗6个月。复发者再用仍有效。约半数患者不能耐受氮芥溶液,可通过降低浓度,或加入1%氢化可的松提高其耐受性。氮芥亦可配成软膏,每日外用,软膏的耐受性较水溶液好,但疗效稍差。
- 卡莫司汀(carmustine,bischloroethylnitrosourea,BCNU) 通常配制成浓度为2mg/mL的酒精溶液,每日1次外用,平均8~12周即可出现明显疗效。本药耐受性较氮芥好,但部分患者可出现骨髓抑制,故治疗期间每月需进行全血细胞计数。另外,还可出现持久而严重的毛细血管扩张。因而该疗法主要应用于不能耐受或对氮芥过敏的患者。
- 贝扎罗汀凝胶 该药是新型外用维A酸类药物,属RXR选择性维A酸(retinoid X receptor-selective retinoids,rexinoids)。其耐受性良好,已于2000年被FDA批准用于ⅠA~ⅡA期皮肤T细胞淋巴瘤。
- PUVA 补骨脂(甲氧沙林)可被表皮细胞摄取,当被光激活后可形成双功能或单功能DNA加合物(DNA adducts)而起效。UVA穿透性强,对斑片/斑块期MF皮损限局者,完全缓解率高达88%,对斑片/斑块期皮损泛发者,完全缓解率为52%,而对肿瘤期无效。红皮病者对PUVA耐受差。一般每周照射2~4次直至皮损清除。研究表明维持治疗并不能防止疾病复发,故应予以避免以减少接受照射的总剂量。
- UVB 目前多应用窄波UVB治疗,其穿透性弱于UVA,仅适用于ⅠA和ⅠB期患者,其治疗红斑期皮损疗效与PUVA相当,但不良反应较后者小,安全性较好,目前尚无诱发皮肤肿瘤的报道。
- 全身皮肤电子束照射(total skin electron beam therapy,TSEB) MF细胞对射线敏感,因而可被低剂量辐射杀死。但应注意严格掌握剂量,以避免对正常组织的损害,照射深度一般控制在5mm以内,通常在8~10周内累积剂量为30~36Gy。虽然其对各期均有效,且有效率高、缓解期长,但不良反应限制了其应用。常见不良反应包括:红斑、水肿、皮损加重、秃发、甲脱落。持久性色素沉着或皮肤干燥也很常见,并可诱发皮肤肿瘤。
- 浅层X线照射 局部浅层X线照射是对少量皮损进行姑息治疗的有效方法,其剂量为10~30Gy,可分次进行。
系统治疗
化学治疗 蕈样肉芽肿对化学治疗相当抵抗,缓解期短。这可能与肿瘤细胞增殖率低,p53灭活突变率高所致的肿瘤细胞凋亡抵抗等有关。因此,对早期MF(包括ⅠA,ⅠB,ⅡA)均应尽量避免应用化学治疗。单药物化疗(如甲氨蝶呤、吉西他滨、苯丁酸氮芥、脂质体多柔比星等)可用于治疗中晚期MF,其中以甲氨蝶呤最为常用。联合化疗(如CHOP方案)常加剧患者的免疫失衡,降低抗肿瘤免疫应答,目前已较少应用。
生物反应调节剂(biological response modifiers)
- 干扰素 干扰素α是Ⅰ型干扰素,可与多种肿瘤细胞表面Ⅰ型干扰素受体结合。有研究表明干扰素α的剂量与效应并无直接相关性,治疗的最佳剂量是每次300万U,每周3次皮下注射。流感样症状是干扰素最常见,并可导致停止治疗的不良反应,因其与剂量呈正相关,可通过减少干扰素用量而缓解。
- 维A酸类药物(retinoids) 本类药物为维生素A的衍生物,可调节肿瘤细胞的分化、诱导肿瘤细胞凋亡,也可能参与了对单个核细胞皮肤浸润的免疫调节,并具有一定的免疫佐剂效应。目前常用阿维A治疗MF,可单独应用,也可与PUVA或干扰素α联合应用。
- RXR选择性维A酸(rexinoids) 贝扎罗汀可高度选择性结合RXR,从而抑制肿瘤细胞生长,并诱导其凋亡。该药已被美国FDA批准用于晚期MF的治疗,也可用于常规治疗无效的早期MF。但其不良反应严重且频率高(如:严重甲状腺功能低下发生率为40%,高甘油三酯血症发生率为79%等),应予注意。
- 地尼白介素(denileukin diftitox) 本药是包含白喉毒素与IL-2的融合蛋白,可选择性结合高亲和力IL-2受体,白喉毒素被细胞内摄,从而导致后者蛋白合成抑制而死亡。肿瘤T细胞表达CD25(IL-2Rα亚单位)时疗效好,故应用该药前应对肿瘤细胞CD25表达情况进行检测。其主要不良反应为“血管泄漏综合征”,表现为高血压、水肿、低白蛋白血症,发生率约25%,用药前应用糖皮质激素不仅可预防该严重不良反应的发生,还可提高地尼白介素的疗效。
其他治疗
- 免疫治疗 阿来组单抗(alemtuzumab)是一种人源化的抗CD52单克隆抗体。CD52表达于正常及恶性的B细胞和T细胞,而不表达于髓系和红系骨髓细胞。阿来组单抗可清除包括肿瘤T细胞在内的淋巴细胞,其不良反应包括机会性感染、严重的心脏毒性等。
- 体外光化学治疗(extracorporeal photoimmunotherapy) 该方法将外周血白细胞分离后,在体外与8-MOP混合,然后照射紫外线。通常每4周连用2天,连续至多6个月。该方法通常耐受良好,主要适用于Ⅲ期MF或Sézary综合征患者,联合应用干扰素α可提高其疗效。
- 中医中药有条件时可结合中医辨证施治,如中医辨证本病早期为血热有毒,后期为久病体虚、血热兼淤,治疗时可考虑在早期用清热解毒,到后期则可用扶正固本、活血化淤等治则。
