
颅咽管瘤(craniopharyngioma)是发生于鞍上区并常累及下丘脑的先天性良性肿瘤。在影响下丘脑的肿瘤中,首先是垂体瘤,其次为颅咽管瘤。后者占先天性颅内肿瘤的60%,占全年龄组颅内肿瘤的4.7%~6.5%,在儿童鞍区肿瘤中,颅咽管瘤约占60%,而儿童颅咽管瘤约占儿童颅内肿瘤的5%(成人3.5%)。
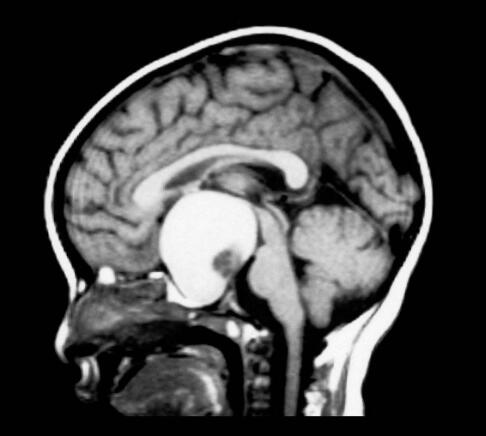
颅咽管瘤的起源仍有争论,主要有两种理论:第一种是胚胎起源理论,第二种是组织化生理论。故先后被称为Rathke瘤、垂体管瘤、颅咽管囊肿、Erdheim瘤、釉质瘤、表皮瘤、垂体柄肿瘤和髓样癌等。
颅咽管瘤可能为二重起源
儿童型肿瘤中心为成釉细胞进行性生长,其中含有栅栏样柱状包绕的胚胎芽苞(成釉型颅咽管瘤),可能是胚胎起源的肿瘤。正常胚胎发育时,Rathke囊与原始口腔相连接的细长管道称为颅咽管,此管随胚胎发育而逐渐退化。Erdheim等认为,颅咽管瘤起源于Rathke囊袋前壁的残余部分、前叶结节部及退化的颅咽管残存鳞状上皮细胞,其好发部位在腺垂体结节部。临床发现,颅咽管瘤常沿Rathke囊袋的发育途径生长,见于第三脑室前端和鞍底,少数位于鞍内、蝶窦甚至鼻咽腔后壁。但也有人认为,颅咽管瘤由垂体组织转化而来。
颅咽管瘤分为釉质细胞瘤样瘤(adamantinomatous tumor)与鳞状乳头样瘤(squamous-papillary tumor)两种类型,后者可能起源于颅咽管的残存组织,伴有腺垂体细胞的鳞状化生、蝶上表皮样囊肿或Rathke囊肿。纤毛状颅咽管瘤(ciliated craniopharyngioma)为Rathke囊肿向鳞状乳头样瘤的转型期肿瘤。成人型颅咽管瘤多由发育成熟的多层鳞状上皮构成,可能由出生后器官转化而成。颅咽管瘤恶性变少见,文献中仅有数例报道。
分型有助于指导治疗和预后判断
解剖分型
根据肿瘤发生的部位,颅咽管瘤可分为5型:
- 鞍内型颅咽管瘤:肿瘤局限于鞍内,但可向邻近部位侵犯;
- 后视交叉型颅咽管瘤:肿瘤位于视交叉和视束之间,可侵及第三脑室;
- Les formes geantes型颅咽管瘤:大肿瘤侵及视交叉前后区,重者侵入幕下或侧裂;
- 非典型型颅咽管瘤:发生于咽部、后颅窝或蝶窦,或侵及松果体区;
- 异位颅咽管瘤(ectopic craniopharyngioma):少见,可位于第四脑室、脑实质等部位。
病理分型
80%~90%的儿童垂体瘤为颅咽管瘤,发病年龄集中在5~14岁。部分患者的细胞单克隆增生与染色体1q、12q、17q或β-连环蛋白(β-catenin)突变有关。根据组织学起源,一般分为牙釉质型、鳞形乳头型和梭形细胞型3型。WHO将颅咽管瘤分为釉质上皮型和鳞形乳头型,另有部分学者认为存在过渡型和混合型。绝大多数肿瘤为前两种类型,其中以牙釉质型多见,主要发生于儿童,多有钙化,最外层是柱状上皮细胞,向心逐渐移行为外层呈栅栏状、内层细胞排列疏松的星形细胞。儿童型颅咽管瘤以囊性(囊性颅咽管瘤,cystic craniopharyngiomas)多见。鳞形乳头型由分化良好的鳞状上皮细胞组成,此型多为实质性肿瘤,成人多见。梭形细胞型属于低度恶性肿瘤。一些良性颅咽管瘤经放射治疗后可转为恶性颅咽管瘤或转为其他类型的恶性肿瘤。偶尔,颅咽管瘤组织含有神经内分泌细胞,可鉴定出神经肽类激素,但一般不引起相应的临床症状。
颅咽管瘤并发症引起多种症状表现
本病以20岁前儿童或青少年多见,但成年人甚至老龄者也可发生。儿童和成人的颅咽管瘤表现有一定差异。儿童以颅内压增高和生长发育障碍为主,成人以视野缺损和垂体功能减退为主。颅咽管瘤的常见并发症有:
- 脑积水和脑萎缩;
- 垂体功能减退症;
- 视神经萎缩和视野缺损;
- 海绵窦症状群;
- 小脑症状群、神经精神症状群;
- 术后下丘脑性肥胖;
- 放射治疗后出现新肿瘤,如树突细胞瘤(astrocytoma)、神经节瘤(glioma)等。
颅内压增高
由于肿瘤压迫第三脑室,堵塞脑室系统引起颅内压增高,表现为头痛、恶心和呕吐、畏光、眩晕、展神经麻痹;严重者可有意识障碍、视乳头水肿、脑积水和继发性脑萎缩与视神经萎缩,有时可引起化学性脑膜炎和蛛网膜炎。儿童有颅缝分离、头颅增大,婴儿有前囟膨隆及头皮静脉怒张等表现。
内分泌功能障碍
71%的患者在首次获得诊断时有内分泌症状。垂体功能减退可能由于肿瘤直接侵犯垂体引起垂体激素分泌不足,或由于肿瘤侵犯下丘脑致促垂体激素分泌不足,或由于肿瘤压迫垂体门脉系统使下丘脑激素不能到达垂体,因而垂体激素分泌低下,产生相应的临床表现。最常见的是GH及性激素分泌减少,患者常有性发育障碍和身材矮小。绝经期前妇女均有原发性或继发性闭经;大部分成年男性可有低促性腺激素性性腺功能减退。儿童患者血GH降低,生长迟缓,约10%患儿出现矮小症伴性发育不全;部分患者的血ACTH、LH、FSH或TSH下降,但临床症状少见,TSH不足引起的继发性甲状腺功能减退约见于1/4患者。极少数患者有腺垂体功能亢进的表现,如性早熟、肢端肥大、泌乳、Cushing病和皮肤色素加深等。合并GnRH瘤时可伴有性腺功能紊乱表现。
下丘脑功能紊乱
视肿瘤压迫情况而出现不同表现,典型者可出现下丘脑功能紊乱的一系列症状,如嗜睡、厌食、消瘦、贪食、肥胖、多汗、汗闭、低血压、发热和体温过低等,AVP下降可导致尿崩症,发生率约20%;GnRH下降引起性腺功能减退和情感与行为障碍。
视力障碍
在成人中易被发现,而在儿童中可能被忽视(误为近视)。此外,可有视野缺损、偏盲。双侧颞下象限性盲说明视交叉后肿瘤压迫视束,双侧颞上象限性盲提示视交叉受压,双眼同向偏盲指向一侧视束受压。严重患者可因原发或继发性视神经萎缩导致失明。肿瘤向鞍旁扩展引起Ⅲ、Ⅳ、Ⅴ、Ⅵ脑神经功能障碍,患者出现复视、动眼神经麻痹、三叉神经痛等;向两侧生长,侵入颞叶,可引起颞叶癫痫;肿瘤向下扩展,侵及脑脚,可产生痉挛性偏瘫,甚至出现去大脑强直状态;肿瘤向前颅窝生长可产生精神症状、癫痫发作、记忆力减退、定向力差和幻味觉等,肿瘤向后生长可产生脑干症状,肿瘤向后颅窝生长可引起小脑症状群。
内分泌功能障碍
临床上可出现GH缺乏(75%)、LH/FSH缺乏(40%)、ACTH缺乏(25%)、垂体柄被压和尿崩症(9%~17%)。
