
英文:Laurence-Moon-Biedl syndrome;
同义名:劳-穆-毕综合征、Laurence-Moon综合征(MIM 24500)、Biedl病、Biemond综合征、Bardet-Biedl综合征、Laurence-Biedl综合征。性幼稚-色素性视网膜炎-多指(趾)畸形综合征、视网膜变性-肥胖-多指综合征、肥胖生殖无能综合征。
溯源与发展
1864年有人报道一家系2例,有视网膜变性、多指症、智力低下患者,但未引起重视。1866年Lanrence和Moon报道一家系4例兄妹患病,具有色素性视网膜病变、智力低下、痉挛性瘫和性腺发育不良表现,后称为Lanrence-Moon综合征(LMS)。
1920年Barded和1922年Biedl分别报道了具有多指、肥胖、智力低下、色素性视网膜病变和性腺发育不良为特点的家系,后称为Barded-Biedi syndrome(BBS)。
1925年Soliscohen和Weiss结合自己发现的4例进行综合分析,命为劳一穆一毕综合征(LMBS)或称Lanrence-Moon-Barded-Biedl综合征(LMBBS),在国内至今沿习应用。
1987年范建华根据最近文献资料,结合1978年McKusick和1985年Cantani等意见,正式提出LMS和BBS是两种独立的综合征,而认为LMBS和LMBBS是人为的错误命名。尽管LMS和BBS临床表现有差异,发病率亦不同,但两者间存有共同的临床表现。1992年白馨芝等有66例的综合报道。我们按历史沿用LMBBS的命名,在临床表现中加以区分。
发病机制
有人推测本病的病因在于基因异常,下丘脑-垂体先天性功能缺陷导致促性腺激素分泌不足,而且合并其他先天畸形,但确切病因及发病机理尚不清楚。
- 病理:尸检可见患者脑及丘脑下部血管发育障碍和胶质细胞减少,灰白结节神经核减少,垂体偶见嗜碱细胞增多,性腺发育不良,原始卵泡或曲细精管减少,间质细胞减少以及精子形成障碍。但上述病理改变尚不足以解释全部临床表现。
- 遗传学:经家系调查和系谱分析,本病遗传方式属AR。以男性患者为多见,男女之比约为3:1~2:1,其机制不详。尚有少数病例可见性染色体非正倍体。基因携带者频率为0.0025。国内66例中有22例(33%)为近亲婚配子女.33例(50%)有家族史,故应禁止近亲婚配,如已生一病儿,第二胎有25%再发危险性。
- 发病率:在欧美文献报道已超过1000例,日本报道300例以上,中国统计报道约近百例(1992-2007年的不完全统计已报道191例以上)。
临床表现
根据临床表现可将LMBBS分成两种类型:
- LMS(劳穆综合征),以小脑共济失调、痉挛性截瘫、眼球震颤多见,约占1/5。
- BBS(巴毕综合征),以多指为多见约占4/5。BBS可分为8种亚型(BBS1、BBS2、BBS3、BBS4、BBS5、BBS6、BBS7、BBS8),累及的基因定位在20p12、16q21、15q23、14q32.1、11q13、4q27、3q13、2q31,这表明BBS的病因是很复杂的。
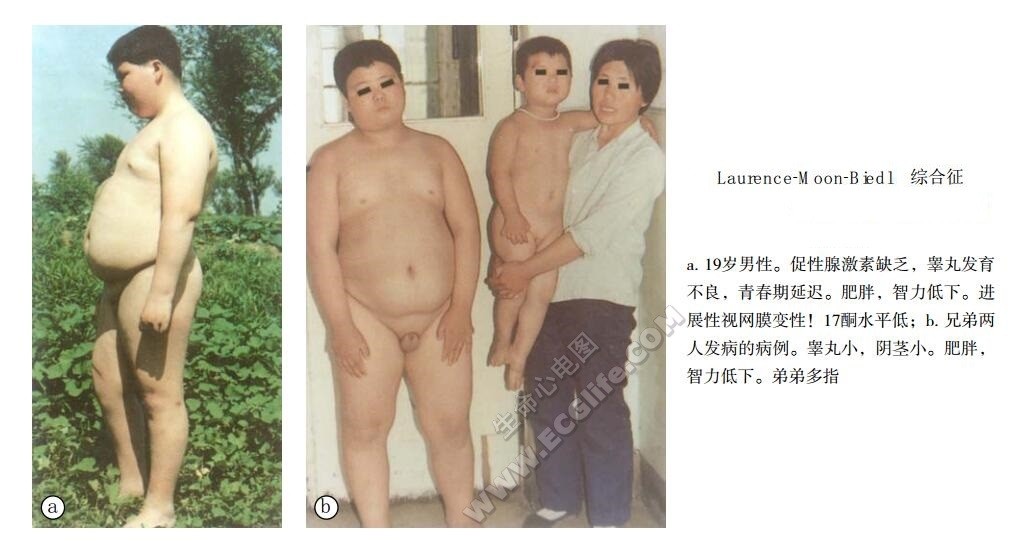
1966年Kleln等将LMBBS分为四型,即1型为完全型;2型为不完全型;3型为非典型型;4型为广泛型。除四项重要主征外,还可以合并其他先天性异常病变。
主要表现为色素性视网膜病变、肥胖、智力低下和性腺发育不良。前两种症状在LMS占60%,BBS占80%~90%;眼球震颤LMS发病率高,多指BBS发病率高。而小脑共济失调(90%)和痉挛性截瘫仅在LMS出现,进行性神经性耳聋和糖尿病出现率不高,不是两病的主要表现,单纯性葡萄糖耐量轻度降低,可能继发于肥胖。
LMS和BBS共有色素性视网膜病变、肥胖、智力低下和性腺发育不良四大主征,不同者为LMS眼球震颤和共济失调发生率高,而BBS中多指发生率高,而且BBS的色素视网膜病变为锥体束营养不良,可引起早期中枢性视力丧失,约60%BBS患者在20岁以后发生真性全盲。EBS常并发肾病和尿毒症而引起死亡。
患者智力低下,出现低智能型面容,眼裂小。有的患者不会写字,也不会做算术。行动比较迟缓,在疾病早期其生活还可以自理。随着病变加重,生活自理能力逐步减退。
由于色素性视网膜病变,导致视力减退,夜间走路多有困难,有时白天也要家属陪扶,不少患者视力在0.01~0.2范围或更差,最后导致仅有光感或失明。
肥胖也是主要体征之一,并出现继发单纯性糖耐量降低现象。
性腺功能减退常见,男性的睾丸容量小于1ml。
BBS常并发肾病,肾脏功能受到明显损害,因此经常进行肾功能检测是非常重要的观察指标,要防止尿毒症。肝纤维化病变也常有发生,部分患者出现嗅觉功能障碍。
- 心血管损害:约为30%。国内心血管发生率为7.5%,有房间隔缺损,室间隔缺损,法洛四联症,大血管转位及单心室等。
诊断、鉴别
- 诊断:Blumel等依据阳性家族史和临床主要表现个别出现,房室阻滞,视力减退、弱智、肥胖和性腺发育异常,再加上多指(趾),六项中具有四项即可诊断。针对不同的临床表现和各自高发生率可区分LMS,BBS。
- 鉴别:需与单纯性肥胖,家族性多指(趾)症鉴别。
治疗及预后
对症治疗。BBS预后不良,应加强治疗视网膜病变,防止失明,积极治疗肾功能衰竭,以免死亡。
