
紫外线对皮肤的急性效应
皮肤光生物反应是以紫外线辐射能量被皮肤特异性的分子或色基(chromophore)吸收开始的,引起重要的生物分子如DNA和蛋白质的直接光化学损伤或间接的氧化损伤。紫外线还可直接作用于表皮中的角质形成细胞、朗格汉斯细胞和真皮中的成纤维细胞等的细胞膜受体,启动细胞信号转导通路,诱导特定基因的表达,最终引起多种细胞内蛋白激酶、各种细胞因子和基质金属蛋白酶的表达与活化。表皮内包含了吸收紫外线光谱的色基,包括核酸、尿刊酸、芳香氨基酸、黑素前体等。DNA的损伤和修复,细胞内信号转导通路的启动,在紫外线辐射后产生细胞因子和炎症介质中起着重要作用。
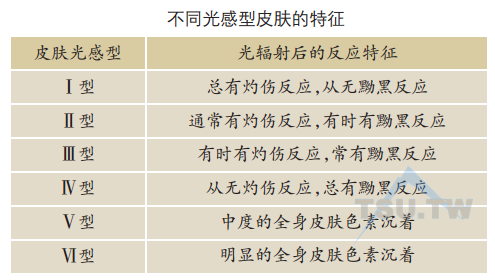
紫外线辐射后皮肤的急性损伤包括日晒红斑和黝黑反应,还有一些其他的影响:局部皮肤和系统免疫抑制,角质层、表皮和真皮增厚,合成维生素D。紫外线辐射引起皮肤的慢性损伤主要是光老化和皮肤癌。根据个体对紫外线辐射后的灼伤反应和黝黑反应的程度,将皮肤分为六个光感型。个体的光感型是由基因决定的。根据对紫外线的反应,Fitzpatrick将皮肤从浅色到深色分为Ⅰ、Ⅱ、Ⅲ、Ⅳ、Ⅴ、Ⅵ六型,分型依据见表下表。
紫外线辐射对皮肤的急性效应主要包括以下几方面。
日晒红斑
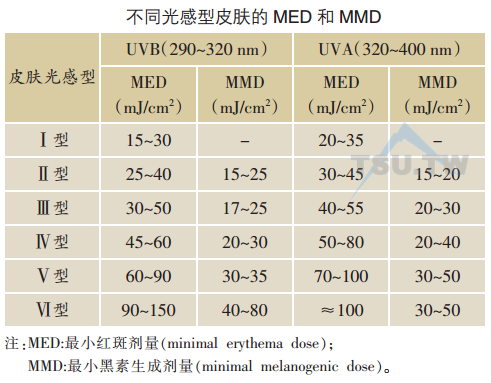
日晒红斑是紫外线辐射后最显著的皮肤急性损伤,对于肤色浅者更为明显。一般红斑程度与辐射剂量相关,紫外线的辐射剂量以焦/厘米2(J/cm2)为单位。紫外线辐射后24小时能诱发皮肤产生刚好能观察到红斑的最小辐射剂量叫做最小红斑剂量(minimal erythema dose,MED)。UVC的MED最小,在0.01 J/cm2以下,UVB和UVA的MED随着波长的增加而明显增加,对Ⅱ型和Ⅲ型皮肤UVB的MED在0.2~0.8 J/cm2之间,而UVA的MED则在1 J/cm2以上。
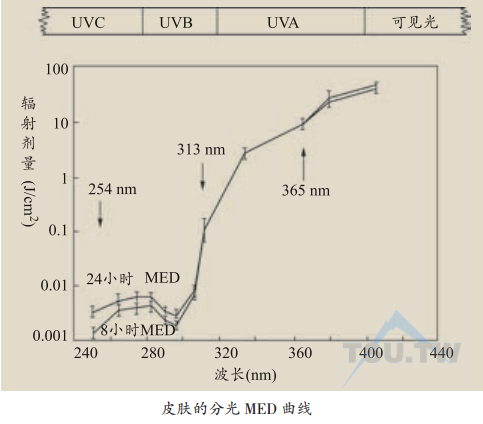
UVB辐射后的红斑反应表现为双相性,即刻反应在辐射后数秒和1秒内出现短暂的潮红,延迟红斑的高峰出现在数分钟和数小时后。即刻反应通常只出现在Ⅰ、Ⅱ型皮肤,延迟反应根据UVB辐射剂量的不同出现在6-12小时。红斑消退需要一天左右,与随后产生的皮肤脱屑、黝黑反应相关。大剂量的紫外线辐射产生更持久和快速的反应。对于肤色浅者和老年人,红斑反应可以持续数周。UVA辐射后出现即刻红斑反应,也有一些研究发现UVA辐射出现双相红斑反应,在辐射后6~12小时延迟红斑出现。老年人由UVA辐射产生的红斑存在更持久。红斑效应随着紫外线波长的增加迅速下降,产生相同红斑的UVA剂量是UVB的1 000倍。日晒红斑随着UVB或UVA辐射剂量的增加而增强,并且与个体差异、身体的部位、环境因素等有关。
日晒红斑的产生可能主要是由于紫外线辐射直接损伤DNA或间接引起内源性的氧化产生光敏反应,进而释放一些化学物质,如组胺、前列腺素、缓激肽,以及细胞因子IL-1、6、8、10、12和黏附分子等。
即刻黝黑和迟发黝黑
即刻黝黑是指皮肤在20~120 kJ/cm2的UVA辐射后数秒钟内开始,持续几分钟到几小时的辐射局部皮肤颜色变灰、变深的反应。较多发生在有色人种中。可能有部分短波可见光也参与这一反应。这可能是由于表皮黑素细胞中的黑素氧化后颜色加深,并重新分布。尽管即刻黝黑并没有起到明确的光保护作用,但可在表皮的基底细胞细胞核上出现有很重要保护价值的“黑素帽”。
迟发黝黑作用是皮肤在紫外线辐射后局部皮肤持续几个星期到几个月的黝黑。这一反应开始于紫外线辐射后的几小时到几天。引起迟发黝黑作用的紫外线辐射剂量因波长不同而不同:较为广谱的UVB为0.04 J/cm2(对于白皮肤的个体此剂量略大于MED);广谱的UVA为15-20 J/cm2(略小于MED);UVC为0.01 J/cm2(大约等于MED)。迟发黝黑的产生可能是被紫外线辐射损伤的黑素细胞中的DNA核菅酸残基激活了酪氨酸酶的活性,促进了新的黑素的产生,并同时将这些黑素通过树状突转运到邻近的角质形成细胞中。黑素细胞的大小和树枝状突起都增加,酶的活性也增加。另外,静止状态的黑素细胞也被激活,细胞分裂增加了新的黑素细胞。但在此阶段白种人对光的敏感性反而降低2-4倍或更多。
增生
UVB或UVC辐射后皮肤的增生从辐射后的几小时到几天开始,可持续1-2个月。UVA 一般没有该效应。皮肤的增生是由于紫外线辐射诱发细胞的有丝分裂,DNA、RNA和蛋白质的合成增加的结果。这种细胞增生的原因还不清楚,紫外线辐射引起的DNA损伤的修复可能是刺激皮肤增生的启动因素。临床上,可以看到病人的表皮、真皮,特别是角质层增厚2~4倍。紫外线辐射诱导的这种表皮及真皮的增厚,可以使皮肤对日晒伤的光保护作用增加10~20倍。
维生素D的合成
维生素D的合成是紫外线辐射皮肤后的重要生理作用。中等剂量的UVB辐射,可迅速地将表皮中的7-脱氢胆固醇转变为维生素D3的前体,并在几天中渐渐地异构为维生素D3,然后通过与血浆中的维生素D结合蛋白结合,进入血液循环。
组织学改变
紫外线辐射后30分钟就可以在显微镜下观察到皮肤组织学改变,UVB辐射可引起表皮和真皮的改变,UVA辐射主要引起真皮的改变。
UVB辐射后30分钟内表皮出现日晒伤细胞(sunburn cell).24小时达到高峰。日晒伤细胞来源于紫外线损伤的角质形成细胞,类似于凋亡细胞。低剂量的UVB辐射还引起表皮朗格汉斯细胞减少,大剂量UVB则引起细胞内损伤。UVA辐射主要引起表皮细胞内水肿。
在UVB和UVA辐射后真皮也改变。辐射后24小时内肥大细胞脱颗粒并迅速恢复正常,血管内皮细胞肿胀,血管周围单核细胞、T淋巴细胞、中性粒细胞浸润,如果此后不再受到辐射,这些改变在1-2周内恢复正常。
紫外线对皮肤的慢性效应
紫外线辐射对皮肤的慢性影响主要是产生光老化和皮肤癌。
皮肤光老化
皮肤老化是由遗传因素(决定自然老化)和环境因素作用所致。环境因素中最重要的是来源于日光的紫外线辐射,紫外线辐射导致皮肤的老化称为皮肤光老化。
光老化的临床和组织学改变:皮肤光老化表现为中老年人日光暴露部位皮肤出现的外观和组织学变化。在外观上表现为:皮肤粗糙、松弛、下垂,出现皱纹、不规则色斑和毛细血管扩张,并可能出现各种良性和恶性肿瘤。上述变化的严重程度取决于个体对日光的耐受性和对日光损伤的修复能力。光老化在曾接受过度日光照射的浅肤色人群中更为显著,在人体面部、颈部及上肢的伸侧部位最明显。
皮肤光老化组织学表现为:表皮在不同部位可出现严重的萎缩或增生,角质形成细胞和黑素细胞都可出现一定程度的核异型。角质形成细胞缺乏分化成熟的有序性,黑素细胞不规则地分布在基底膜上方,朗格汉斯细胞数量有明显减少,白种人光暴露部位皮肤朗格汉斯细胞数量仅为遮盖部位的50%左右。光老化皮肤最明显的变化表现在真皮细胞外基质,最显著的是日光性弹性纤维病变,在HE染色切片中表现为大量与弹性纤维染色相似的物质聚集,免疫组化研究显示这些物质由弹力蛋白等弹性纤维的成分构成,但是这些物质的排列不似正常弹性纤维那样有序。同时还会出现胶原纤维数量和结构的变化,研究表明在日光性弹性纤维病变的部位出现胶原纤维的降解,胶原水平减少伴异常弹性纤维沉积是皮肤日光损害的特征。光老化部位皮肤小静脉由于血管壁明显增厚而出现血管屈曲扩张,另外由于血管周围支撑结缔组织的减少和血管内皮细胞的损伤而出现表浅小血管扩张。皮肤附属器在组织学上的变化表现为毛囊扩张,萎缩的皮脂腺存在于弹性纤维变性的真皮中,这些表现与毛囊失去结缔组织支持有关。
皮肤光老化的作用光谱:UVB和UVA辐射均可与皮肤光老化有关。UVB主要作用于表皮角质形成细胞和黑素细胞,不仅损伤DNA,而且产生可溶性的因子进一步作用于真皮。UVA辐射直接影响表皮和真皮。
虽然UVA的生物学活性不如UVB,但由于日光中UVA的剂量可以是UVB的10~1 000倍(根据季节和每天的时间变化),并且能够透人皮肤深层,因此目前认为在光老化的病理机制中,UVA和UVB起着同样重要的作用。
皮肤光老化机制:皮肤光老化发生的确切机制仍不明确,但已有大量研究从分子水平揭示了导致光老化的多种路径。
- 端粒在皮肤光老化中的作用端粒在染色体末端,由“5TTAGGG3”片断重复串联成一环形结构。紫外线辐射造成急性DNA损伤,端粒环形结构破坏,3’端TTAGGG暴露,被感受器蛋白识别,从而发动DNA损伤后的信号。这一结果也促进了细胞分裂端粒严重变短和环形结构紧缩时发生的自然老化。
- 线粒体DNA(mitochondrial DNA,mtDNA)突变与光老化 mtDNA的突变频率是细胞核DNA突变频率的50倍,而mtDNA的修复功能有限,因此mcDNA突变后线粒体功能下降是引起自然老化的一个重要原因。研究发现,老化的皮肤内线粒体基因组突变增加,在老化皮肤内,4 977bp mtDNA缺失(又称为共同缺失)比光保护部位内增加10倍。而且在mtDNA突变的基因敲除小鼠中发现,mtDNA突变是引起老化的重要原因。此外,还发现在UVA诱导mtDNA突变增加的同时,氧耗量降低,线粒体膜电位、三磷酸腺苷(adenosine triphosphate,ATP)含量和基质金属蛋白酶-1(matrix metalloproteinase,MMP-1)增加,而MMP的组织特异性抑制物(tissue-specific inhibitors,TIMP)没有改变。这也提示了mtDNA突变与光老化有关。
- 光老化中基质金属蛋白酶和胶原合成的改变紫外线作用于人体皮肤后通过产生的活性氧、细胞表面的生长因子受体、细胞因子受体及许多酶的激活,可迅速诱导c-Jun蛋白的表达,c-Jun和c-Fos组成异二聚体,与其他蛋白(包括NF-κB)一起组成激活转录因子激活蛋白(AP-1),AP-1可调控基质金属蛋白酶(MMP)的表达,MMP是一个锌依赖性的酶家族,能够特异性地降解细胞外基质,最终导致皮肤光老化。MMP家族中至少有4个成员由AP-1调节:胶原酶、92kD明胶酶、基质降解酶和金属弹力蛋白酶,紫外线照射入体皮肤可诱导这4种MMP表达,与紫外线诱导AP-1一致,紫外线照射8~16小时后即可出现MMP的表达,并持续24小时以上。UVB和UVA均可引起MMP表达增加,但TIMPs并没有改变。此外,胶原合成减少,其机制尚不清楚。
光致癌
光致癌中紫外线的波长和剂量:大量的实验动物研究表明紫外线辐射可以引起皮肤癌,肿瘤的类型与动物种类、辐射剂量、皮肤类型、皮肤厚度等有关。
UVB辐射的致癌作用是UVA的1 000倍,随着波长的增加,紫外线致癌作用减弱,但UVA并非没有致癌作用,剂量足够大时同样可以导致皮肤癌。由于地面日光中的紫外线主要是UVA,UVA的致癌作用不容忽视。研究发现UVA并不是皮肤癌发生的启动因素,但是UVA有着重要的促进作用。
多次小剂量紫外线辐射比减少次数的大剂量紫外线辐射导致皮肤癌的可能性更大,减少紫外线辐射次数可以推迟皮肤癌的发生。非致癌剂量的紫外线在皮肤反复接触致癌促进剂情况下可以导致皮肤癌发生。
光致癌中的免疫因素:实验研究发现紫外线辐射诱导小鼠发生的皮肤肿瘤移植到正常同源受体后不再继续生长,如果在移植前用紫外线辐射宿主,则移植的肿瘤就会继续生长。进一步研究表明抑制性T淋巴细胞导致紫外线辐射后小鼠对肿瘤没有排斥反应。将小鼠的某一部位进行小剂量的紫外线辐射,然后在另一部位照射致癌剂量的紫外线,皮肤癌的发生率大大提高。这些研究均表明紫外线辐射皮肤导致系统免疫抑制,在紫外线诱导皮肤癌发生中起着重要作用。
光致癌的遗传因素:许多肿瘤是由于控制细胞生长的基因发生突变所致。紫外线诱导DNA损伤的修复基因突变可导致紫外线相关的皮肤癌发生,DNA损伤修复基因在紫外线引起的皮肤癌过程中起着重要作用。原癌基因突变也导致肿瘤发生,在紫外线诱导的皮肤癌中60%-100%出现p53基因突变,长期接受紫外线辐射未出现皮肤癌之前就可以检测到这种突变。10%~40%皮肤癌出现ras基因突变,这些基因与细胞膜和细胞核间的信号转导有关,突变激活了ras基因,产生持续信号转导,细胞生长失去控制。另一个与皮肤癌相关的基因是谷胱甘肽转移酶1(glutathione transferase,GSTM1),它的缺失使基底细胞癌和鳞癌的发生率增加。
光致癌的分子机制:紫外线致癌与化学致癌物有着许多共同点,如可以诱导增生、刺激生成可以促进肿瘤形成的酶、诱导炎症反应等。紫外线辐射引起的免疫抑制也与肿瘤形成相关。此外紫外线辐射还可以改变局部环境,从而促进肿瘤生长。而且紫外线辐射导致的这些改变也有利于其他致癌物如X线、病毒、化学致癌物等诱导产生皮肤癌。因而紫外线辐射除了直接引起皮肤癌外,还是其他皮肤致癌剂的辅助因素。
紫外线辐射致癌的分子机制不是十分清楚,并不是所有的都与紫外线引起的DNA损伤有关。紫外线辐射可产生多种DNA损伤,其中最主要的是环丁烷嘧啶二聚体(cyclobutane pyrimidine dipolymer,CPD)和6-4光产物( photoproduct,6-4PP),两者都可导致突变,其他损伤包括单链断裂、DNA交联、嘌呤光产物等。这些DNA损伤在致癌过程中的作用取决于细胞在这些损害永久性整合入基因组之前的修复情况,人体细胞可以采用切除修复机制高效率地修复DNA损伤,但是修复机制受到多个基因控制,而这些基因本身也可能成为紫外线损伤的目标,因此DNA修复和细胞周期控制可能失败。DNA损伤还可能是由于紫外线辐射产生的单线氧、超氧离子和过氧化物所致。对皮肤癌p53基因分析发现,CPD和6-4PP是与皮肤癌形成相关的主要突变。多数p53突变是C、T转换和CC、TT的突变,这些突变只在UVB、UVC辐射后产生,UVA辐射和化学致癌物均不能使p53产生这样的突变。长波紫外线光化学疗法(PUVA)引起皮肤癌的p53突变主要发生在5’-TA/5’-TAT或其邻近部位。
紫外线辐射与黑素瘤:紫外线辐射诱导皮肤黑素瘤的产生机制不是十分清楚,只有恶性雀斑样痣与紫外线辐射相关,其他黑素瘤并没有更倾向于发生在身体的暴露部位,也没有随着年龄的增长而增加发生率。对小鼠进行紫外线辐射没有能诱导出黑素瘤,但紫外线辐射与肿瘤促进剂或化学致癌物共同作用可诱导小鼠产生黑素瘤。此外,紫外线辐射可以促进辐射部位皮肤产生原发或转移的黑素瘤,这与紫外线辐射表皮产生黑素瘤生长因子和降低皮肤免疫反应有关。
尽管如此,仍有一些证据间接表明皮肤黑素瘤的发生与紫外线辐射有关,但黑素瘤很少发生p53突变,紫外线辐射可能仅仅是引起黑素瘤发生的许多因素之一。
