
Kussmaul和Maier在1866年首先提出结节性动脉周围炎(periarteritis nodosa)这个病名。1903年Ferrari认为该病有多血管的累及,动脉壁全层的炎症。因此,命名为结节性多动脉炎。本病为主要侵犯小、中等大肌性动脉的节段性坏死性血管炎,而不侵犯静脉或淋巴管,血清ANCA一般阴性。有两种类型:良性皮肤型和系统型。多数有系统性病变,20%~25%的病例仅表现有皮肤症状。可因血管炎而发生缺血、梗塞和出血,出现终末器官的损伤。好发于中年男性,男性发病率是女性的4倍,平均发病年龄是45岁。年发病率是46~4.9/100万,发病无人种差别。
病因及发病机制
病因尚未明确,可能与以下因素有关:
- 感染:患者常有潜在的链球菌、乙肝或丙肝病毒感染。5%~54%患者与乙肝病毒的感染有关。其他感染包括链球菌(尤其是儿童)、结核杆菌、细小病毒组B19、HIV。
- 其他相关疾病:本病可伴随于包括SLE、炎症性肠病、毛细胞白血病、家族性地中海热、大动脉炎、复发性多软骨炎、Cogan病和静脉吸毒等。
发生机制可能是免疫复合物沉积引起的Ⅲ型变态反应,还可能与动脉分枝点的剪应力(sheer stress)增加,造成内皮细胞炎症因子的上调有关。在这些剪应力部位的内膜中有很多的巨噬细胞,这使此动脉分枝点的内皮细胞易受炎症的伤害。有的学者认为系统型有免疫学机制的参与,而皮肤型无免疫学机制的参与。与系统型不同,皮肤型不累及小动脉的分叉部位。
临床症状
结节性多动脉炎因累及不同部位的血管炎而症状及体征有多样性。
皮肤型
皮肤结节性多动脉炎主要局限于皮肤,无内脏累及,常有复发性皮肤、关节、肌肉和神经累及的症状,良性病程,此型是否为结节性多动脉炎的早期表现,还是局限型还有争论。皮肤表现为真皮或皮下结节,最常位于足、踝部附近、小腿,并向心性地发展至大腿、臀部、上肢和手部。偶发于躯干、面、头皮及肩。两侧发生,但不对称。直径0.5~2cm,一般比其他下肢结节性疾病的结节小,数目不定。结节单个或成群发生,成群的结节多在网状青斑处发生。结节硬,易触到,表面淡红色或鲜红色,常有触痛、压痛及自发痛。可沿血管发生,持续1周或更久而消失。由于侵犯局部血管,局部组织缺血,产生淤斑、坏死或溃疡,其边缘不整,周围常有网状青斑围绕。可有肌痛,特别在夜间显著。
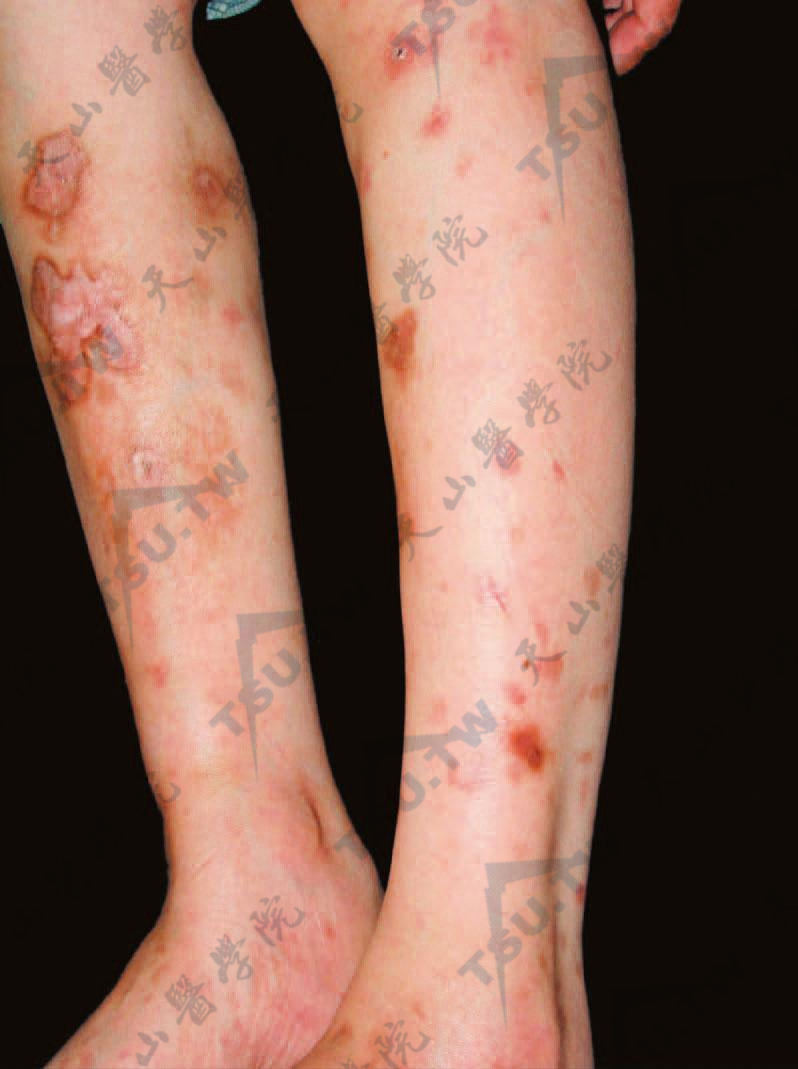
小腿皮下结节,多发。部分皮损可形成溃疡,周围有网状青斑围绕
亦可侵犯肢体的周围神经而表现为轻度压痛、麻木或轻度麻痹。有的还有关节疼痛、关节周围肿胀及红斑。在足及小腿踝部都可见网状青斑。
系统型
40%~60%的病例有皮肤损害,可有多种损害。一般认为系统型的皮肤症状与皮肤型没有区别。但皮疹急性发生,有出血、大疱、急性栓塞及溃疡,表现为明显急性炎症。最具诊断价值的损害是直径5~10mm的皮下结节(15%的病例),单个或成群分布,常沿血管分布。结节上方的表皮正常或有轻微的红斑。这些结节常有疼痛,并可有搏动性。常发生于下肢,尤其是膝以下。也可发生淤斑和指(趾)的周围性的坏疽。也可发生网状青斑、大疱、丘疹、猩红热样损害和荨麻疹样损害,或深在性的软组织肿瘤样的肿块。
可累及全身所有的中小动脉。全身不适、乏力、高血压、心动过速、发热、水肿、体重减轻是该病的主要表现。肝大、黄疸、淋巴结肿大、血尿和白细胞升高也是常见的表现。也可有关节痛、心肌梗死、心包炎、心包积血、充血性心力衰竭、急性主动脉炎、肾小球硬化。常表现为足下垂的多发性单神经炎,是本病的重要体征。累及脑膜、脊柱和颈动脉时,可导致偏瘫和抽搐及各种出血症状,亦可发生周围神经炎;有的可发生视盘水肿、视神经萎缩、角膜炎、角膜溃疡及巩膜结节;肠道受累则发生剧烈腹痛、便血、肠梗死等症状;可有睾丸炎;肺和脾脏极少累及。
本病病程不定,通常6个月至1年,但有些病例反复发作可达数年。系统型结节性多动脉炎不治疗5年存活期只有13%;常死于肾衰竭或心血管或胃肠道并发症。皮肤型预后较好,皮损可持续数年。系统型积极应用糖皮质激素和(或)加用免疫抑制剂治疗后,预后明显好转,存活率可达90%以上。当有肾小球硬化、高血压及侵犯腹部者,预后较差。有25%死于肾衰竭、脑或腹部出血、心肌梗死、心力衰竭或反复感染。
血常规检查可有进行性正细胞性贫血,白细胞计数升高,有的可高达40×109/L,70%的病例中性粒细胞增多,常有嗜酸性粒细胞增多,可有血小板增多,但亦可发生白细胞减少及相对淋巴细胞增多。血沉快。C反应蛋白升高。可有高球蛋白血症(包括巨球蛋白血症和冷球蛋白血症)。蛋白电泳γ球蛋白及α2球蛋白增高,白蛋白减少。类风湿因子可阳性,有些病人有低补体血症。侵犯肾脏时则有蛋白尿、血尿及管型(70%的病例)。应做丙肝的血清学检查。系统型患者血清中抗中性粒细胞胞质抗体(C-ANCA)可阳性,动脉造影可显示肾脏、肝脏和内脏血管有多发性的中等大动脉的动脉瘤性扩张。皮肤型与系统型从实验室检查及病理检查结果均表示为同一疾病的两种表现,其主要区别见下表。
皮肤型及系统型结节性多动脉炎比较表

组织病理
真皮与皮下组织交界处及皮下组织的中、小动脉的炎症性坏死性闭塞性全层动脉炎,伴有灶性脂膜炎改变。根据皮损发展的不同时期,动脉的变化可分4期:
- 第1期为变性期,中膜及外膜形成坏死灶,有纤维素样坏死,很快扩展至内膜,坏死部位可形成小动脉瘤,破裂时血管周围有出血。
- 第2期为炎症期,在动脉坏死区内及其周围有致密的炎细胞浸润,主要为中性粒细胞,亦可有嗜酸性粒细胞及单核细胞,受累的动脉腔可形成血栓。
- 第3期为肉芽肿形成期,即坏死的血管壁被肉芽组织所代替,内膜增生使管腔部分甚至全部闭塞。
- 第4期为纤维化期,即毁坏的血管壁被纤维组织所代替,管腔缩小、闭塞或再通。部分病例直接免疫荧光可见C3和IgM沉积于血管壁。
诊断及鉴别
本病的临床表现多样。诊断主要依据:
- 特征性的皮肤损害,皮下结节及网状青斑;
- 常有多系统受累,有发热、体重下降、乏力、肌痛、关节痛等;
- 多项实验室检查异常,如白细胞增高,嗜中性粒细胞升高、血小板增多、正细胞性贫血、血沉快,C反应蛋白增高、血清γ球蛋白增高、HBsAg阳性、高丙种球蛋白血症、冷球蛋白血症及血尿、蛋白尿、管型尿等;
- 皮肤、肌肉、神经、肾组织等活检示中小动脉的炎症、坏死性和阻塞性全动脉炎。血管造影术发现有血管壁的动脉瘤性扩张也有助于诊断该病。P-ANCA阳性率只有20%。
结节性多动脉炎和显微镜下多血管炎的临床鉴别见下表。
结节性多动脉炎和显微镜下多血管炎的临床比较

治疗
系统型
全身用糖皮质激素仍是主要的治疗方法,可改善5年存活率,从10%~13%升到48%~57%。每日使用泼尼松或泼尼松龙60~100mg[1~2mg/(kg·d)],症状改善后,逐渐降至维持量约每日10~20mg。经3~6个月的治疗后,病情缓解,可逐渐减量至停用。环磷酰胺可与糖皮质激素合并应用或有时单独使用,开始的单次剂量为2mg/(kg·d),对于严重的病例可增加剂量,用药期间应监测血常规,当病情稳定至少1年时,可以减少环磷酰胺的量并停用。平均需用18~24个月。血浆置换联合IFN-α2和(或)阿糖腺苷对伴乙肝病毒感染的结节性多动脉炎治疗有效。
也有报道单用抗病毒药治疗有效的。对于乙肝相关的结节性多动脉炎,糖皮质激素只能在开始时短期治疗,因为糖皮质激素会造成乙肝病毒的复制。对系统性症状应对症治疗。
皮肤型
非甾体抗炎药对皮肤型的症状治疗有效。有些病例需大剂量糖皮质激素,以后逐渐减量。常用青霉素治疗和预防儿童皮肤型结节性多动脉炎,因为儿童皮肤型结节性多动脉炎的发病与链球菌感染密切相关。其他的治疗包括双嘧达莫、磺胺吡啶、己酮可可碱和氨苯砜。有报道每周小剂量的甲氨蝶呤(每周7.5mg~20mg)对有些糖皮质激素系统或皮损内注射无效的病例有效。个案病例报道小剂量甲氨蝶呤和穿弹力袜对于大部分病例可有效。
有报道1例皮肤型结节性多动脉炎,用泼尼松、硫唑嘌呤、环磷酰胺等传统治疗无效的病例,给予静脉注射丙种球蛋白每月1g/kg,3个月可获满意疗效。
