-
吉特林综合征(Gitlin Syndrome)
本病为性联重症联合型免疫缺陷病[severe combined immunodeficiencv(sex-linked type)]。病变基因位于X染色体上,为X连锁隐性遗传,患者多为男性。发病机制为IL-2、IL-4、IL-7受体&
16 -
维斯科特-奥尔德里奇综合征
本病为X连锁隐性遗传,有免疫异常伴有血小板减少和湿疹。血小板减少是由于患者的血小板生存期显著地缩短,而骨髓中的巨核细胞计数是正常的。患者的血小板比正常小,常有各种畸形,
17 -
软骨-毛发发育不良综合征
软骨-毛发发育不良综合征(Cartilage-hair Hypoplasia Syndrome)病为常染色体隐性遗传,其病因是由于RMRP基因突变,导致线粒体RNA合成代谢障碍所致。Gatti等(1969年)报道2例短肢侏儒
19 -
奥门综合征
奥门综合征(Omenn Syndrome)又名家族性网状内皮细胞增生症伴嗜酸性粒细胞增多(familial reticuloendotheliosis with eosinophilia)。本病为常染色体隐性遗传,因参与T淋巴细胞受
20 -
共济失调-毛细血管扩张症(易斯巴尔综合征)
共济失调-毛细血管扩张症(Ataxia Telangiectasia)又名路易斯-巴尔综合征(Louis-Bar syndrome)伴有共济失调和毛细血管扩张的免疫缺陷病,是一种常染色体隐性遗传的原发性免疫缺陷
21 -
胸腺瘤伴免疫缺陷病(Good综合征)
胸腺瘤伴免疫缺陷病(Thymoma with Immunodeficiency)又名Good综合征(Good's syndrome),是一种少见的、成年发病的综合征,以胸腺瘤、低丙种球蛋白血症、低外周血B淋巴细胞和伴有T淋
22 -
慢性肉芽肿病
出生后不久即发病,包括全身性皮肤的反复顽固的感染,大致可分为白细胞趋化功能不全综合征(dyschemotactic syndrome)、吞噬功能障碍综合征(dysphagocytotic syndrome)、抗体缺陷综
24 -
高免疫球蛋白E综合征(乔布综合征、HIES)
本病又名乔布综合征(Job syndrome),其特点为异位性皮炎样损害,血清IgE增高,中性粒细胞趋化性障碍及反复发生化脓性感染。本病多为散发,少数为常染色体隐性或显性遗传。1966年Davis
25 -
白细胞黏附分子缺乏症
病因:常染色体隐性遗传,β2整合素基因突变导致中性粒细胞表面CD18分子表达障碍,不能形成黏附分子受体,粒细胞趋化性运动能力的降低,最终影响吞噬功能。临床症状本病的主要特
26 -
谢迪埃克-赫加希综合征(CHS、先天性白细胞颗粒异常)
谢迪埃克-赫加希综合征(Chediak-Higashi Syndrome)又称先天性白细胞颗粒异常综合征、溶酶体病、非霍奇金淋巴瘤,1943年由Besuez等首次报道。1952年和1954年Chediak及Higashi分
27 -
髓过氧化物酶缺乏症
髓过氧化物酶(MPO)为含有一个α大亚单位和一个β小亚单位的与铁相结合的复合体,存在于中性粒细胞和单核细胞中,可催化H2O2和卤化物反应生成高效氧化剂。其主要的基因位
28 -
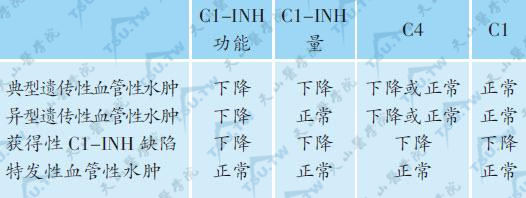
遗传性血管性水肿(C1酯酶抑制剂缺乏)
遗传性血管性水肿(HAE)又称C1酯酶抑制剂(complement 1 esterase inhibitor,C1-INH)缺乏症,为常染色体显性遗传病,多数有家族史,但亦有约20%的患者无家族阳性病史可寻。其特点为反复发
29 -
C1/C2/C3/C4缺陷
这是遗传性补体成分缺陷的疾病:C1缺陷可以是其亚单位C1q、C1r、C1s的缺陷。几乎每个补体成分的缺陷均有合并某一种或多种结缔组织病的报道,特别是系统性红斑狼疮。广泛的C2缺
30