
一般原发性骨质疏松症的遗传因素的影响占70%~80%,有些病例存在明显的家族发病倾向,且其性质未明,但不属于家族性骨质疏松的范畴。家族性骨质疏松(familial osteoporosis)是指骨质疏松具有强烈的家族发病特点,且遗传病因相对明确或已经明确,以BMD明显降低和易于骨折为特征的一组临床综合征。如果把病因明确(多为单基因点突变所致)的遗传性代谢性骨病所伴有的骨质疏松称为继发性家族性骨质疏松症的话,那么病因尚未完全明了者可称为原发性(或特发性)家族性骨质疏松症。
原发性家族性骨质疏松症主要与遗传因素相关
Henderson等报道,对69例原发性骨质疏松病例的421个一级亲属和748个二级亲属的遗传学咨询调查结果表明,45%的患者应诊断为家族性骨质疏松症。在这些家族中,母亲患病的危险性为33%,女儿为19%。
Chuvashia等用骨皮质指数(cortical index,CI;即皮质骨厚度/骨直径比值)作为指标,探讨骨量和骨几何结构特征的光线,用复合分离分析法(complex segregation analysis)发现,73%的骨皮质指数是由遗传因素决定的,这一遗传因素(很可能为主导基因)控制了CI的基础水平、CI变化的年龄以及程度。家族性骨质疏松还可能与VD受体(VDR)基因的多态性(BB、TT、AA)有关(母亲-女儿或女儿-女儿相关),但也有反对意见。Deng等报道,用复合分离法发现941例(51个家族)决定BMD/BMC的一个主导遗传位点,这个位点对BMD/BMC的贡献率约为75%,导致BMD/BMC的差异达16%。一过性髋部骨质疏松(transient osteoporosis of hip,TOH)的遗传性状和临床表现具有家族发病特点,主要患病对象为中年男性和妊娠妇女(第3个三月期),主要症状是髋部疼痛和髋部暂时性低骨量,但无髋关节变窄、破坏等征象,一般在数月后自行恢复。而在另一些家族中,似乎父亲-女儿的遗传关系比母亲-女儿更密切。Krall等对40个家族的成员进行BMD测量,发现父母的平均BMD(以Z值表示)与儿子和女儿的BMD值相关,遗传对BMD的贡献率为46%~62%。
目前,流行病学遗传研究的结果可总结为如下几点:
- 正常人群峰值骨量和BMD的高低受遗传因素的控制;
- 遗传因素来源于父母双方,但以母亲对女儿的BMD影响似乎更大;
- 在遗传因素和环境因素的共同作用下,达到峰值骨量后的增龄性骨量丢失的年龄和部位均不相同;
- 老年人的BMD可能主要受环境因素的影响,而青年人的BMD主要受遗传因素的影响;
- BMD降低是导致骨折的重要因素之一,由于BMD受遗传因素的影响,故父母发生骨折,则儿女骨折的风险亦明显增加;
- 体质性生长与青春期发育延迟(constitutional delay of growth and puberty,CDGP)虽然可用生长和青春期发育延迟来解释其所伴有的低骨量与骨质疏松,但这些人在青春发育期以前即存在骨的矿化不良,说明患者的低BMD主要由遗传因素控制;
- 母亲和女儿的绝经年龄密切相关,提示卵巢功能和更年期年龄也主要由遗传因素决定(下表)。
家族性骨质疏松的病因分类
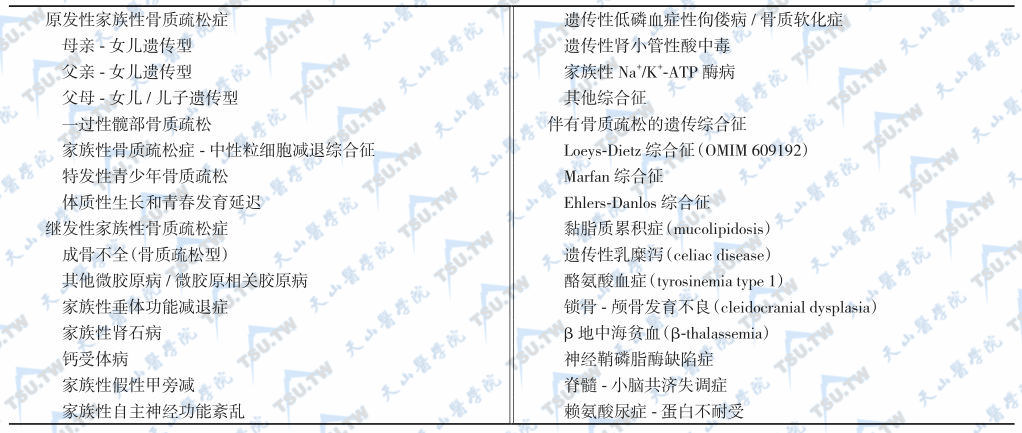
遗传综合征伴发低骨量/骨质疏松症
引起家族性继发性骨质疏松的临床综合征很多,在这些综合征中,骨质疏松仅为患者的表现之一,不典型者常需借助基因诊断来鉴别。家族遗传特点很突出,但骨密度降低有明确的病因。
一、家族性含钙肾石病:家族性含钙肾石病(familial calcium stone disease)的遗传倾向较一般肾石病或一般骨质疏松要明显得多,但其遗传因素至今未明。
二、钙受体病:细胞外液Ca2+浓度的维持和调节依赖于细胞膜上受体(CaR)的作用,CaR基因突变引起两类刚好相反的临床综合征。家族性低尿钙性高钙血症(FHH)和新生儿重症甲旁亢(NSHPT)为CaR基因失活性突变所致;而常染色体显性遗传性家族性低钙血症(ADH)和常染色体显性遗传性甲旁减(ADHPT)为CaR基因活化性突变所致。这些钙受体病均可伴有BMD降低,但钙受体病并非一般意义上的家族性骨质疏松,而是家族性甲旁亢或甲旁减中的特殊类型。
三、家族性假性甲旁减:家族性假性甲旁减(伴骨质疏松)与银屑病存在某种遗传关联,患者还可能伴有Albright骨营养不良症(Albright osteodystrophy)。这些患者除存在假性甲旁减的临床表现外,性腺功能减退、骨质疏松和皮肤病变均较突出,但其关联的遗传因素未明。
四、遗传性低磷血症性佝偻病骨质软化症:特发性成年发作性高磷尿症(idiopathic adult-onset phosphate diabetes)伴有低骨量或骨质疏松,BMD降低。这些患者的肾小管磷最大重吸收率(TmP)与GFR的比值<0.77。虽然一般将本症列为低磷血症性骨质软化症佝偻病/骨质软化症范围内,但早期可仅表现为骨质疏松,故对家族性青少年或成年骨质疏松者必须测定TmPO4/GFR和骨代谢生化指标,排除低磷血症性佝偻病/骨质软化症可能。遗传性低磷血症性佝偻病伴高钙血症(hereditary hypophosphatemic rickets with hypercalcemia)患者的血磷和TmPO4下降,但血钙升高(吸收性),尿钙排出增多。骨病变主要表现为骨质软化症,补磷治疗效果良好。
五、遗传性肾小管性酸中毒:遗传性肾小管性酸中毒可分为多种类型,有时患者以骨质疏松和低BMD而就诊,易与家族性骨质疏松混淆。
六、特发性青少年脊柱侧弯和特发性青少年关节炎:常伴有明显的全身性BMD降低,但因有原发病的突出表现,其鉴别不难。
七、先天性中性粒细胞减少:近年来,发现青少年型骨质疏松与先天性中性粒细胞减少症有关联,外周血中性粒细胞减少,伴严重骨质疏松和反复骨折(家族性骨质疏松-中性粒细胞减少综合征,familial osteoporosis-neutropenia syndrome),其发病机制未明,可能与OPG/RANKL的功能异常有关。
八、家族性渗出性玻璃体-视网膜病:家族性渗出性玻璃体-视网膜病(familial exudative vitreoretinopathy,FEVR)的发病与视网膜血管病变有关,其病因又与LRP5突变关联,同时伴低骨量。
九、家族性下丘脑地高辛缺乏综合征:家族性下丘脑地高辛缺乏综合征(familial hypothalamic digoxin deficiency syndrome)表现为低血压、反复发作的呼吸道感染、精神抑郁、神经性贪食和骨质疏松。血地高辛、HMG-CoA还原酶活性、dolichol和自由基下降,而红细胞Na+/K+-ATP酶和自由基清除酶活性升高。由于Na+/K+-ATP酶活性和血镁升高,血清中酪氨酸分解产物增多而色氨酸分解产物下降,血清GAG分解酶和糖水化酶(glycohydrolases)活性降低(但细胞膜总GAG增多),而胆固醇/磷脂比值下降;血清自由基清除酶活性升高使自由基明显减少。
十、家族性自主神经病:家族性自主神经病(familial dysautonomia)患者有多发性骨折,但骨痛不明显。骨脆性和血NTX增加,骨龄延迟;血BALP降低,BMD和脊柱Z值低。
十一、伴有骨质疏松的其他遗传综合征:几乎所有的遗传性体质性骨病均伴有低骨量/骨质疏松。Hajdu-Cheney综合征(常染色体显性遗传)是一种先天性骨增生不良性疾病,表现为肢端骨溶解,指(趾)缩短甚至消失、颅骨畸形和全身性严重骨质疏松(高转换型伴骨形成障碍)。本病呈家族性发病,母亲和女儿易发病。碱性转化生长因子受体1或2(TGF BR1/ TGFBR2)突变引起Loeys-Dietz综合征(LDS,OMIM # 609192)、Marfan综合征(Marfan syndrome)和Ehlers-Danlos综合征。
