-
马方综合征
本病系由Marfan于1896年首先描述,也称蜘蛛状指(趾)综合征,是一种以特殊性蜘蛛样四肢表现、多器官受累(主要为主动脉扩张、晶体异位和骨骼畸形)的遗传疾病,较罕见。病因是结缔组织代
31 -
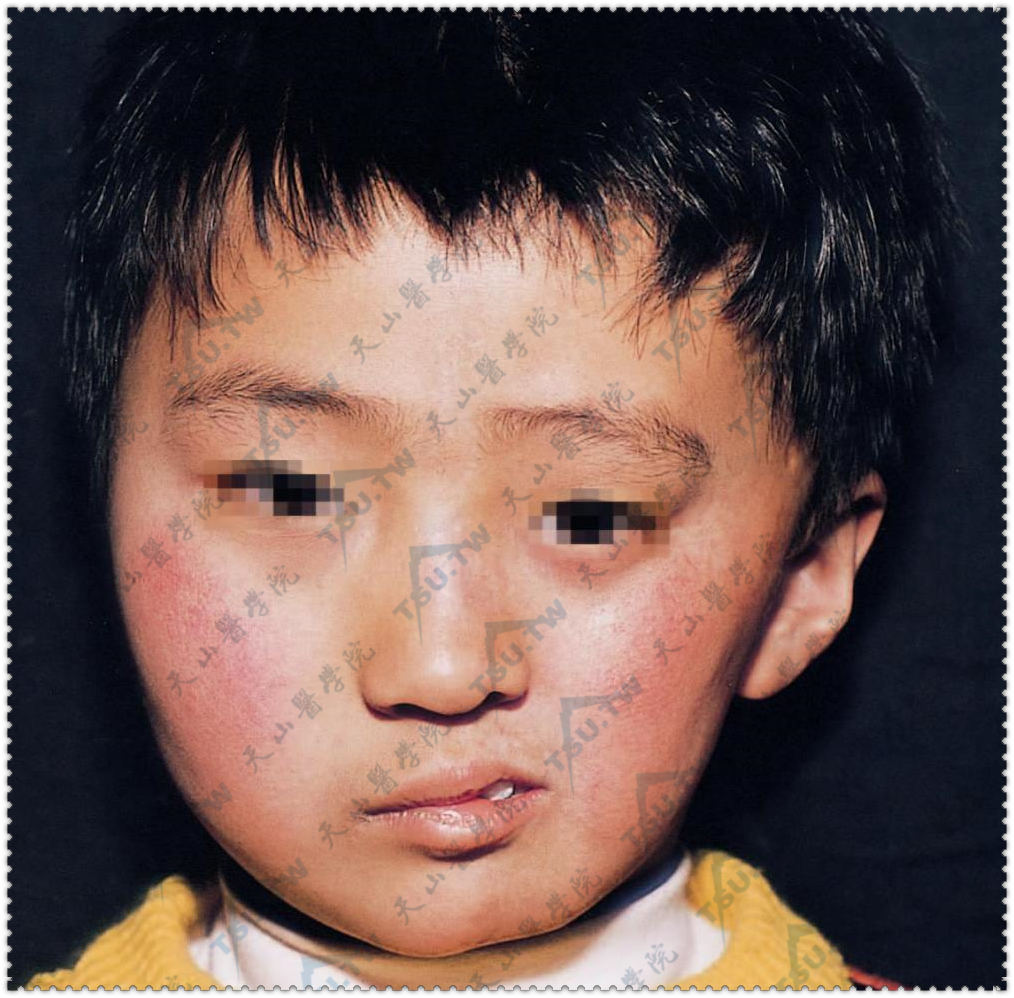
面部偏侧萎缩
本病系指一侧颜面部皮肤、皮下组织、肌肉,甚至骨骼发生进行性萎缩,由Parry于1825年首先描述,又称进行性单侧面萎缩(hemiatrophia facialis progressive)或帕里-龙贝格综合征(Parry
32 -
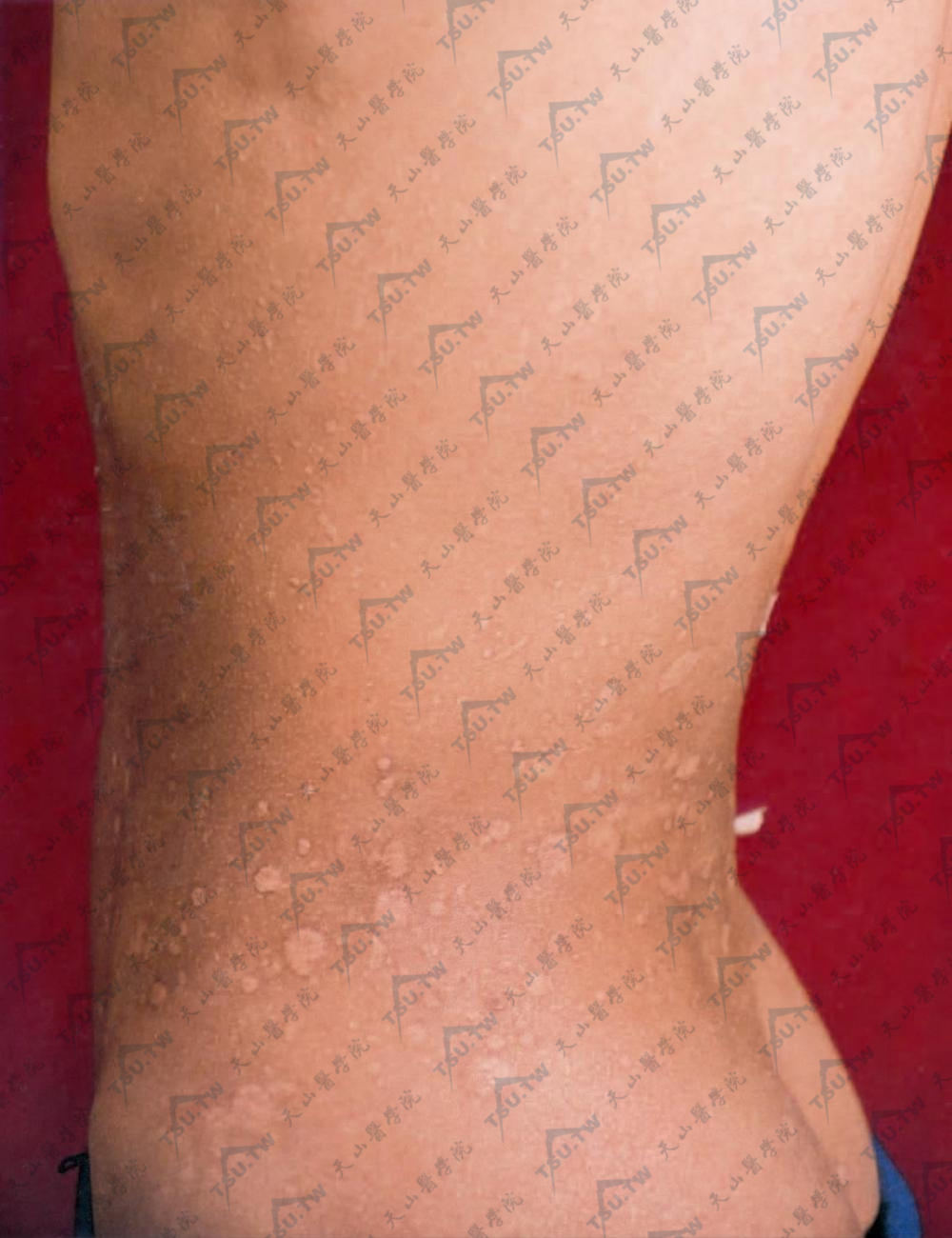
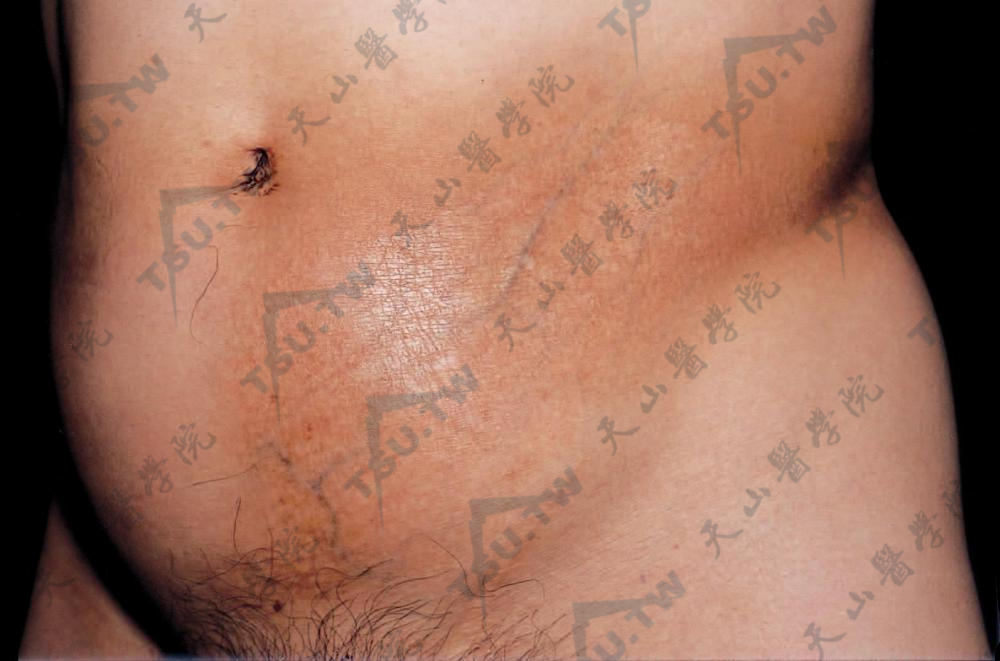
Jadassohn-Pellizari型皮肤松弛症
本病又称红斑性皮肤松弛症(erythematous anetoderma),系由Jadassohn于1891年首先描述,Finger等称其为斑状萎缩性皮炎(dermatitis atrophicans maculosa)。病因:尚不明了,有内分泌(性
34 -
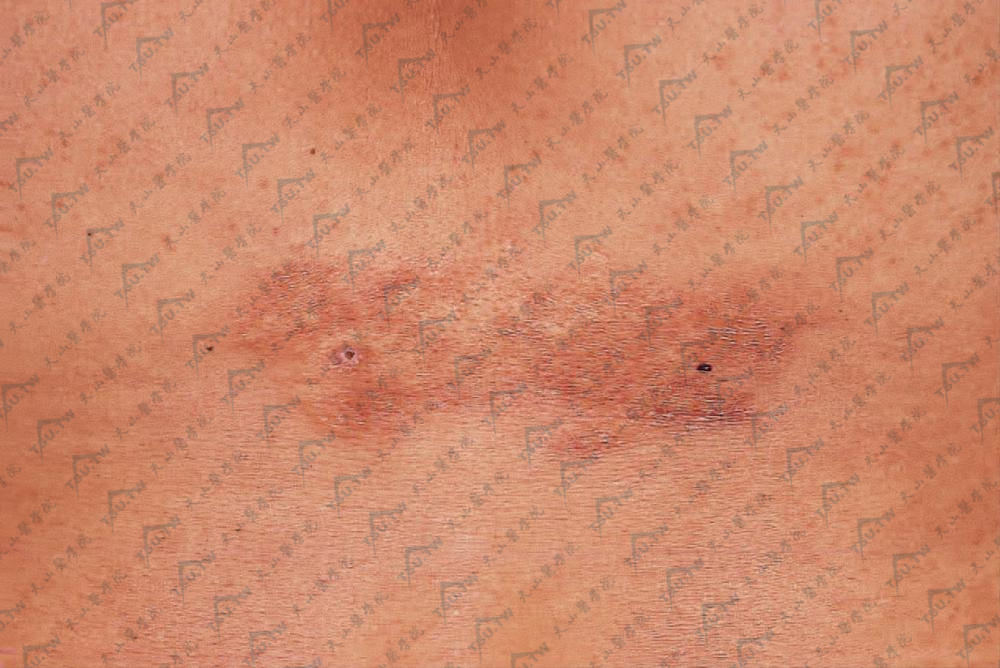
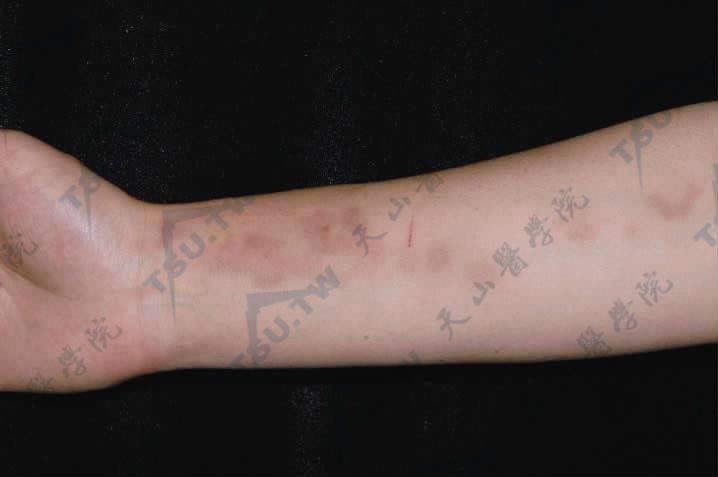
Schweninger-Buzzi型皮肤松弛症
本病又称无红斑性皮肤松弛症或皮肤多发性良性肿瘤样新生物,其特点为临床与组织病理变化始终缺乏炎症反应。病因不明,多见于中年(30~50岁)女性,偶见于男性。初起为躯干及上肢近端突
35 -
皮肤痘疮样斑状萎缩
本病病因不明,往往有家族史(显性遗传),也有些病例可能继发于水痘。症状始于儿童期。皮损好发于面部、胸部和腹部,而四肢末端不会出现皮损,损害均为凹点状皮肤萎缩,呈圆形或卵圆形,孤
36 -
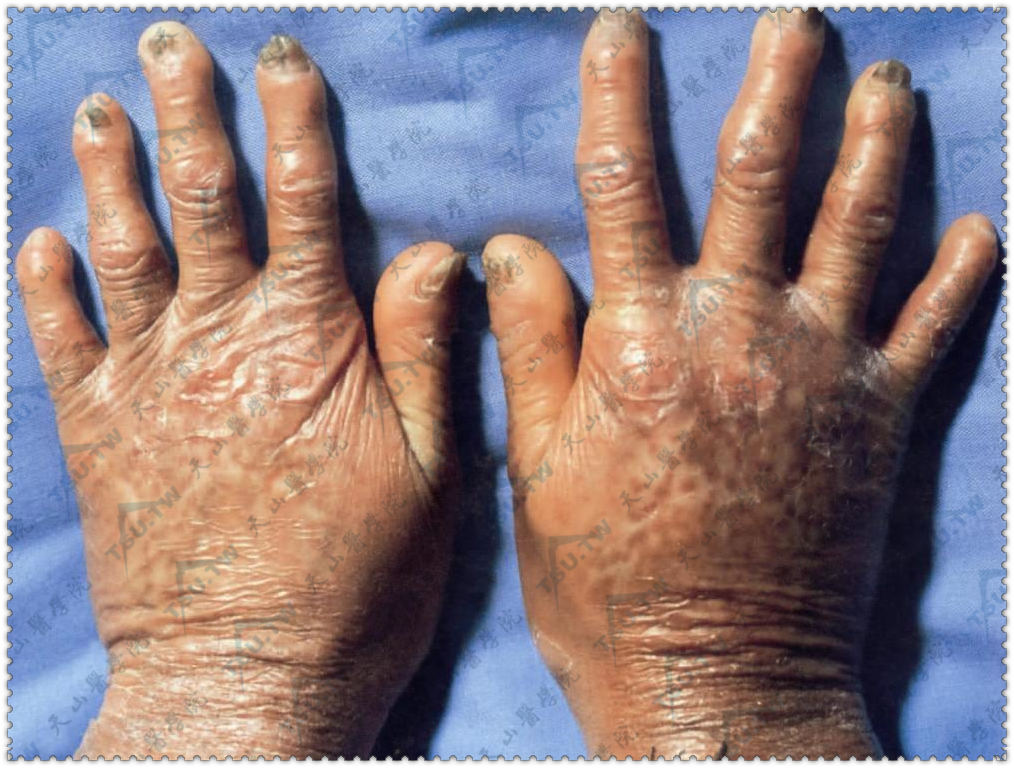
萎缩性慢性肢端皮炎
本病又名原发性弥漫性萎缩,主要发生于欧洲人中,美洲人中也有发病,我国极为罕见。女性发病率高于男性。病因本病是一种螺旋体病,为博氏螺旋体感染的晚期后遗症。Hauser等认为由硬
39 -
毛囊性皮肤萎缩
本病是一种少见的遗传疾病,由Miescher于1943年首先描述。可以单独出现,但主要是作为某些遗传性综合征的组成部分。多在幼年期发病,皮损主要位于手、足背及腿、臂伸侧,也可见于颊
40 -
进行性特发性皮肤萎缩
在1923年,Pasini描述了一种色素性皮肤萎缩的特殊型,其临床上和组织病理上均与局限性硬皮病萎缩期或任何已知的其他萎缩不同;以后又发现了许多病例,主要在阿根廷由Pierini(1935年)
41 -
穆兰线状萎缩性皮病
本病1992年Moulin首次报道,皮损呈带状的色素性萎缩性斑,1994年Baumann命名为Moulin线状萎缩性皮病。本病的病因不明,由于皮损通常沿Blaschko线分布,因此推测可能系胚胎发育早期
42 -
趾(指)断症及假性趾(指)断症、阿洪病
趾(指)断症又称自发性趾(指)脱落症(dactyloysis spontanea)或阿洪病。文献记载多认为是热带地区的一种地方病,多见于非洲热带的成年黑人,尤其是西非海岸的黑人。男性多于女性。本病
43 -
局部全层萎缩
本病又称环状脂肪萎缩,少见,系患者某些部位发生皮肤及皮下脂肪萎缩,有时伴有肌肉、骨骼萎缩或发育不全。病因不明,可能是多种病理过程的结局。本病分为两型。Growers全层萎缩(pan
44 -
萎缩性多软骨炎
本病又名复发性多软骨炎(relapsing polychondritis)、系统性骨软化症(systemic chondromalacia)或冯·迈温伯格-奥尔塞西综合征(von Meyenburg-Althess syndrome)。本病系由
45