-
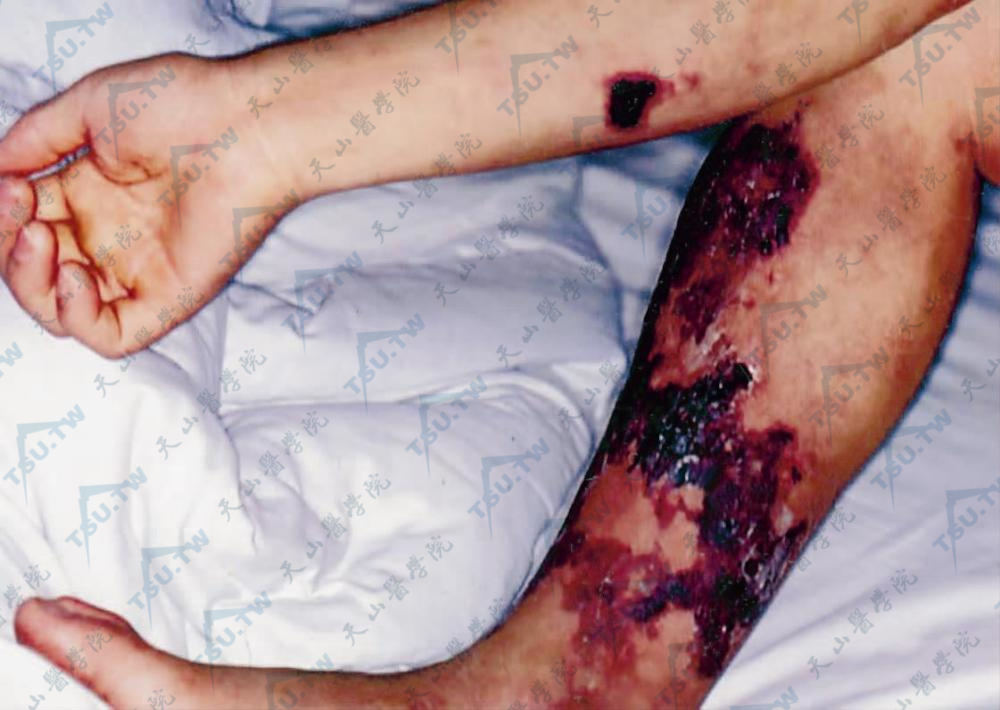
肾衰竭相关性钙化性脂膜炎
慢性肾衰竭患者常有慢性钙磷代谢异常,引起继发性甲状旁腺功能亢进,出现血钙、磷升高,钙沉积于组织中,但钙磷水平正常者,也可出现组织内钙沉着。病因及发病机制肾衰竭引起高甲状旁
17 -
外伤性脂膜炎
本病又称外伤性脂肪坏死(traumatic fat necrosis)。由于外伤引起皮下脂肪坏死,坏死组织可通过破口排出体外,轻微外伤即可发病,患者常不能回忆起此前有外伤史。有时外伤性脂膜炎为
18 -
硬化性脂肪肉芽肿
本病又名油性肉芽肿(oil granuloma)、石蜡瘤(oleogranuloma,或oleoma,或paraffinoma)、化学性脂膜炎(chemical panniculitis)。注射硅酮、矿物油、动物油或植物油后,注射物在皮肤中发
20 -
硬化性脂膜炎
本病1955年Huriez首先报道,又称脂肪皮肤硬化症(lipodermatosclerosis)、皮肤脂肪硬化症(dermatoli-posclerosis)、硬皮病样皮下组织炎(hypodermatitis sclerodermiformis)、静脉淤
21 -
结缔组织性脂膜炎
本病又称脂肪萎缩性脂膜炎(lipoatrophic panniculitis)、自身免疫性脂膜炎(autoimmune panniculitis),发生于儿童或成人,为少见的脂膜炎。以前文献将很多病例归于Weber-Christian
22 -
补体缺陷性脂膜炎
补体缺陷除可并发部分脂肪萎缩综合征外也有报道补体缺陷可发生小叶脂膜炎。Pascual(1987年)报道2例复发性发热性脂膜炎,有获得性补体缺陷(获得性C1抑制剂缺乏)和副蛋白血症(IgG ka
23 -
儿童颅部筋膜炎
Laner et Enzinger(1980年)首先报道9例,命名为儿童颅部筋膜炎,以往曾以不同名称报道。无论临床上或病理上都类似结节性筋膜炎,但本病病变均在头颅部,而且均为儿童患者。本病可作为
25 -
嗜酸性脂膜炎
Burket(1985年)首先报道本病,Winkelmann(1986年)总结18例患者,其特征是皮下脂肪坏死伴显著的嗜酸性粒细胞浸润,主要在脂肪间隔内,也可累及脂肪小叶,并常波及真皮层,与嗜酸性蜂窝织炎(We
26 -
嗜酸性肌痛综合征
本病是与L-色氨酸摄取相关的肌病,有嗜酸性粒细胞增多,肺部病变和皮损。发病机制不明,氨基酸分析发现有称为E峰(peak E)的混杂物,可能在发病中起作用。临床表现皮损多样性,包括局限
27 -
水肿性瘢痕形成血管炎性脂膜炎
本病发生于儿童,是一种新的多系统疾病,死亡率达35.7%。皮肤损害开始类似于种痘样水疱病,但以后累及非曝光部位。皮损发展为深部溃疡及水痘样瘢痕。组织学上有明显的结节性淋巴组
29 -
痛性肥胖病
本病又称德卡姆病(Dercum's disease),为一少见疾患,常发生于肥胖或肥胖的绝经期妇女,患者有精神异常,皮下硬结对称分布于脂肪沉着部位,有剧痛。病因病因不明,该病常见于绝经期妇女,有
30