-
促血小板生成素(TPO)
促血小板生成素(TPO)是c- mp受体的配体,促血小板生成素(TPO)为天然分子结构,经修饰并PEG化后,即为巨核细胞生长发育因子(MGDF)。两种分子的活性相似。后者的稳定性好,半衰期长。促血小
21 -
血小板改变和血栓性疾病
(一)血小板减少和增多在严重细菌、病毒和真菌性败血症,常合并血小板减少。在细菌性败血症患者,2/3以上有不同程度血小板减少,1/3有明显减少(<50×109/L),但除非合并DIC,临床基本
23 -
血小板膜糖蛋白Ⅱb/Ⅲa受体阻滞剂
ADP、胶原、凝血酶、血栓素(TXA2)等激动剂诱发血小板聚集时,分别通过受体占有及信息传递使血小板膜糖蛋白(GP)Ⅱb/Ⅲa形成复合物(纤维蛋白原受体),在膜上表达并发生构象改变。GPⅡb/
24 -
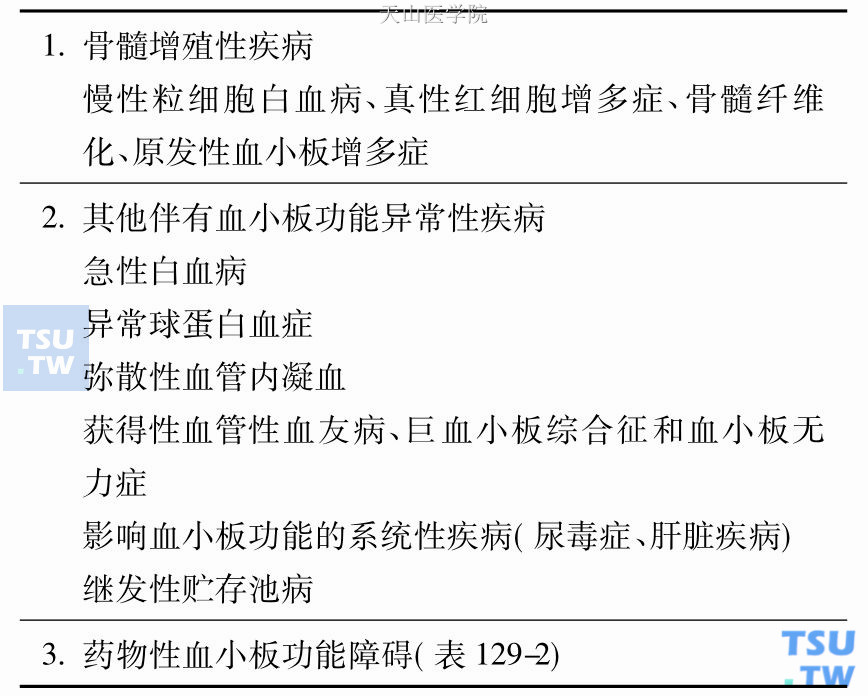
其他伴有血小板功能异常的疾病
急性白血病和骨髓增生异常综合征出血是急性白血病的常见症状,严重者可有消化道、呼吸道出血、颅内出血,是引起死亡的主要原因之一。急性白血病出血的原因是多方面的,最常见的原
26 -
关于继发性血小板功能异常
继发性血小板功能异常(secondary abnormality of platelet function)较先天性血小板功能异常更多见,可继发于多种血液病、非血液病或某些药物(下表),对血小板功能的影响也是多方面
27 -
其他少见的天性血小板功能异常性疾病
(一)Quebec血小板病本病呈常染色体显性遗传。发病机制未明,可能与导致血小板α颗粒内大多数蛋白质降解有关的缺陷相关。患者血小板α颗粒内多聚素(multimerin)异常而导
29 -
血小板贮存池病(α、δ、αδ)
血小板含有特异的α颗粒与致密颗粒,具有重要的功能,缺乏这些颗粒将引起出血。α贮存池缺陷本病又称灰色血小板综合征,由于瑞氏染色血小板呈灰蓝色而命名,属常染色体遗
30 -
MYH9综合征的发病机制与诊断鉴别
发病机制与临床表现MYH9综合征是一类较为常见的常染色体显性遗传的巨大血小板,其临床表现为血小板巨大、血小板减少与中性粒细胞包涵体,部分患者合并有肾炎、耳聋和先天性白内
31 -
血小板无力症的病因、症状及检查
病因和发病机制血小板无力症(Glanzmann’s thrombasthenia,GT)是一种常染色体隐性遗传性出血性疾病,由于血小板膜糖蛋白GPⅡb(αⅡb,CD41)和(或)GPⅢa (β3,CD61)质或量的
32 -
巨大血小板综合征
病因和发病机制巨大血小板综合征(Bernard Soulier syndrome,BSS)是一种罕见的常染色体隐性遗传性出血性疾病,多发生于近亲结婚的家族;其发病机制是血小板膜糖蛋白GPⅠb/Ⅸ/Ⅴ复合
33 -
血小板无效生成性血小板减少
本病的特征为:骨髓巨核细胞数量正常或增多,血小板更新率和产生率明显降低。叶酸、维生素B12缺乏可引起本病,但多伴有贫血或白细胞减少。除血小板无效生成外,约1/3至半数患者尚有
35 -
先天性及遗传性血小板减少症
根据遗传方式可将先天性及遗传性血小板减少症分为三类:一、伴性隐性遗传性血小板减少症 Wiskott- Aldrich综合征。 单纯性(isolated)血小板减少症。二、常染色体显性遗传性血小
36 -
周期性血小板减少症
此病比较少见。自Dammer等1920年首次报道本病以来,至今文献报道的病例仅50余例。国内有个别报道,到目前为止我们共收治了4例。病因不明。本病的特点是周期性发生血小板减少,多
37 -
人类免疫缺陷病毒引起的血小板减少性紫癜
Morris等1982年首次报道健康男性同性恋患者发生自身免疫性血小板减少,以后在静脉药物成瘾者、获得性免疫缺陷综合征(AIDS)及其相关性疾病、血友病患者也发现了类似的变化。血小
38 -
输血后紫癜
输血后紫癜(post- transfusion purpura,PTP)相当少见,最早由van Loghem报道,至今已有200余例报道,国内有少数报道。其特征是输注含血小板成分血液后约1周患者突发血小板减少,其严重
39