-
蛋白C缺乏症
蛋白C(protein C,PC)抗凝系统是由凝血酶调节蛋白(thrombomodulin,TM)、蛋白C(PC)和蛋白C抑制物(PCI)组成。蛋白C是一种由肝脏合成的维生素依赖性酶原蛋白,分子量62kDa,含461个氨基酸,由轻
21 -
输注维生素K依赖性凝血因子浓缩剂导致血栓形成
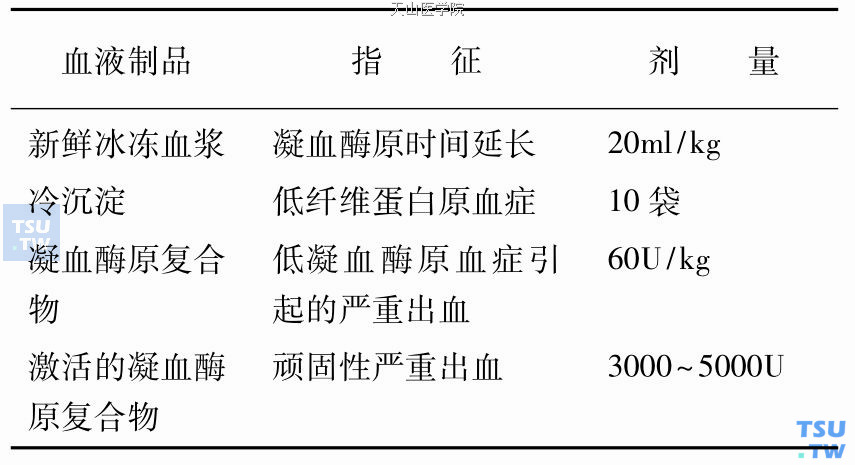
对于先天或获得性凝血因子缺乏症患者以及循环血液中具有FⅧ抗体患者并发出血时,有指针输注冻干的人凝血酶原复合物浓缩剂(PPC)。制剂除有携带肝炎及HIV感染危险外,极少报道对外
22 -
遗传性联合凝血因子缺乏症
遗传性联合凝血因子缺乏症又称家族性多凝血因子缺乏(FMFD)综合征。根据缺乏因子的组合可分为多种类型FMFD。Ⅰ型为FⅤ和FⅫ缺乏;Ⅱ型为FⅧ和FⅨ缺乏;Ⅲ型为FⅡ、FⅦ、FⅨ和FⅩ缺
23 -
遗传性因子Ⅻ、激肽释放酶原、高分子量激肽原缺乏症
凝血因子Ⅻ(FⅫ)激肽释放酶原(PK)和高分子量激肽原(HMWK)与FⅫ一样均被认为是凝血途径中的接触因子。凝血机制内源途径或接触途径始于FⅫ与负电荷表面的作用,形成FⅫa,FⅫa激活FⅪ成
24 -
遗传性因子Ⅺ缺乏症(血友病C)
遗传性因子Ⅺ缺乏症由Rosenthal等首先报道于1953年,当时命名为血友病C,但现在一般称为因子Ⅺ缺乏症。本病多见于犹太人后裔,其杂合子发生率可达2%~13%,纯合子发生率约0. 1%。但在
25 -
遗传性因子Ⅹ缺乏症
本病最早报道于1956年,Hougie和Telfer分别报道1例名叫Stuart的男性病例和1例名叫Prower的女性病例。故因子Ⅹ(FⅩ)又称为Stuart- Prower因子。本病罕见,有人估计发病率低于1/50
26 -
遗传性因子Ⅶ缺乏症
凝血因子Ⅶ(FⅦ)曾称为稳定因子。FⅦ缺乏症首先由Alexander报道于1951年,现已有150例。本病罕见,发病率估计为1/50万。Goodnight 1971年将此病分为FⅦ缺乏症和FⅦ异常血症。前者
27 -
遗传性因子Ⅴ缺乏症(副血友病)
Owern于1947年首先报道此病。此病有时被称为副血友病。在凝血因子Ⅴ(FⅤ)发现前,曾认为有一个加速因子(又称易变因子,曾被命名为凝血因子Ⅵ),以后的研究证明前加速因子和因子Ⅴ为同
28 -
第XⅢ因子抑制物和获得性第XⅢ因子缺乏症
第XⅢ因子抑制物极其罕见,至今约有20例。抗体多为IgG型,绝大多数特异性地作用于第XⅢ因子中a-亚单位的不同部位,并直接抑制第XⅢ因子的活性。其中2例发生在第XⅢ因子缺乏症多次
30 -
先天性第XⅢ因子缺乏症
自1960年瑞士Dukert首例报道,该病为少见的凝血因子缺乏的遗传性疾病,目前认为其发病率为1/100万~300万人,此病呈常染色体隐性遗传,约1/3病例发生在双亲有血缘关系的家庭。病因和
31 -
先天性维生素K依赖性凝血因子缺乏症
1997年德国的Oldenburg等诊断一个患有联合维生素K依赖性凝血因子缺乏症(VKCFD)的家庭,这个家庭来自黎巴嫩,父母为近亲,有2名孩子分别在出生时及1年内死于脑出血,存活的7名子女中有
32 -
其他影响维生素K依赖凝血因子的疾病
淀粉样变性可以引起纤维蛋白原减少症和第Ⅹ因子的减少。虽然引起第Ⅹ因子减少不多见,但获得性第Ⅹ因子缺乏症多数由淀粉样变性引起。引起第Ⅹ因子缺乏原因是淀粉样变性轻链蛋
33 -
食物性维生素K缺乏症
食物性维生素K缺乏发生于各种原因导致长期不能进食的患者,特别是接受全部消化道外全营养并缺乏足够量维生素K补充(约需150μg/d)的患者。当有出血症状且凝血酶原时间延长时应
36 -
维生素K的来源和在凝血因子合成中的作用
维生素K的基本结构为甲萘醌。天然的维生素K有两种,即维生素K1(叶绿醌)和K2(甲基醌),均为脂溶性。前者来源于植物,后者由肠道菌群合成。人工合成的维生素K3(亚硫酸氢钠甲萘醌)和维生素
38 -
获得性因子XⅢ抑制物
因子XⅢ又称为纤维蛋白稳定因子(fibrin stabilizing factor)。在遗传性因子XⅢ缺乏症患者输注治疗后,有可能会出现因子XⅢ抑制物。自发获得性因子XⅢ抑制物极为少见,仅有20例报
39 -
获得性纤维蛋白原、纤维蛋白多聚体抑制物
有报道遗传性无纤维蛋白原血症(hereditary afibrinogenemia)患者输注由正常人血浆制备的纤维蛋白原后,会出现多克隆抗纤维蛋白原抗体。此外,在SLE、单克隆γ球蛋白疾病等患
40