-
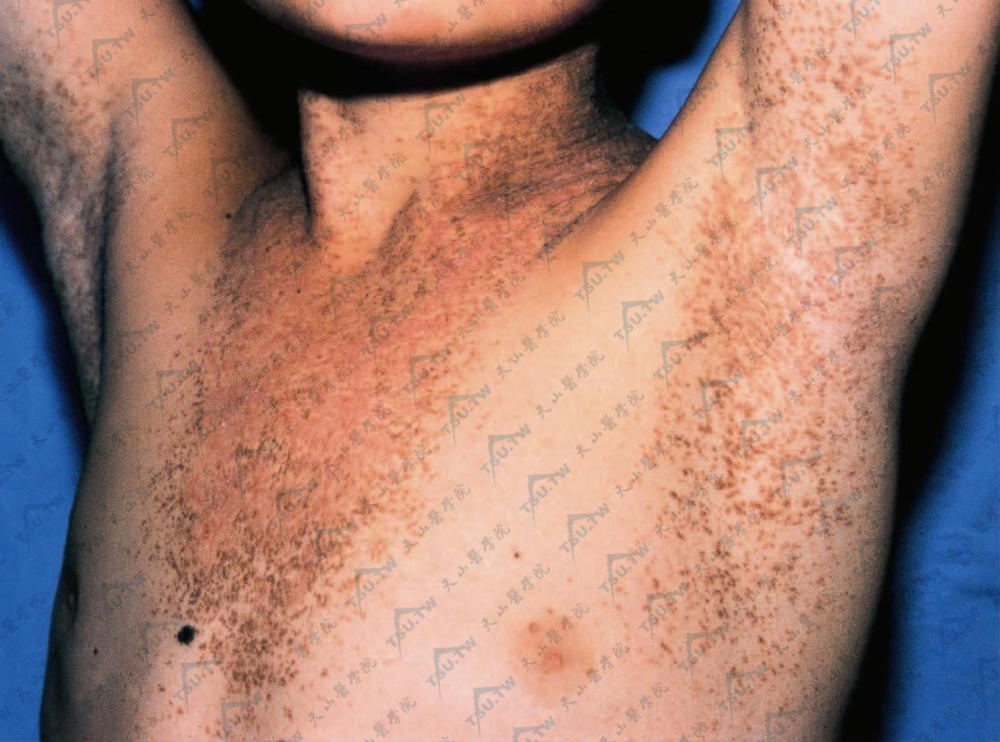
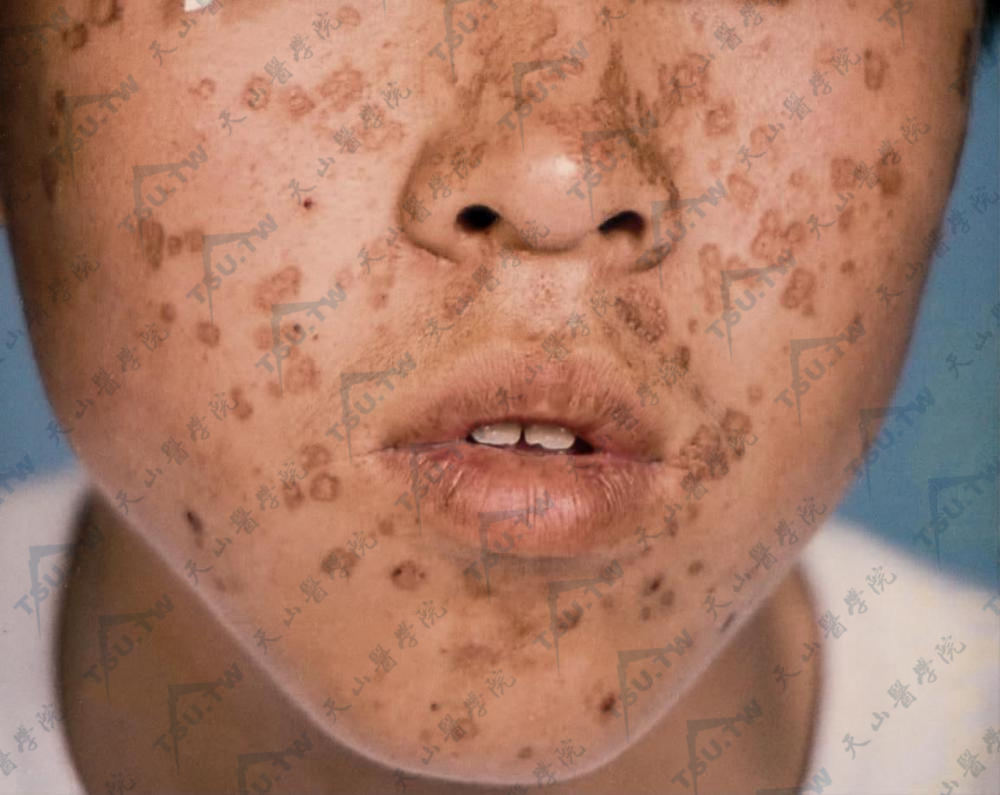
毛囊角化病(毛囊角化不良病)
本病又名假性毛囊角化不良病(dyskeratosis pseudofolliclaris),系1889年首先由Darier命名,故又称达里埃病(Darier's disease)。本病是一种少见的,以表皮细胞角化不良为基本病理变化
1 -
汗孔角化症
汗孔角化症(Porokeratosis of Mibelli)是一种较少见的、起源于遗传的慢性进行性角化不全性皮肤病。以边缘堤状疣状隆起、中央轻度萎缩、组织学上存在角质样板层(cornoid lamell
2 -
播散性浅表性光线性汗孔角化症
本病系1966年由Chernosky首先报道,是汗孔角化症中最常见的一种类型,因有皮损广布于暴露部位,病损表浅播散,发疹与日光有明显关系等临床特点,故有此名。病因及发病机制与Mibelli汗
3 -
传染性毛囊角化病
本病又称流行性痤疮(epidemic acne)、流行性毛囊性发疹(epidemic follicular eruption)、流行性毛囊性角化病(epidemic follicular keratosis)和Brooke病,是与毛囊角化病相似,发生于
5 -
点状汗孔角化症
本病是汗孔角化症的一个新的类型,由Rahbari等在1977年命名。据1988年统计,全世界共报道了8例,其中男性7例、女性1例,年龄从16~64岁不等,约半数在儿童或青春期发疹,多数皮疹表现为1~2
6 -
疏散性跖部汗孔角化症
本病见于成人,女性较多,男女之比为1:4。其特征为边缘锐利、橡胶样、宽基底的丘疹,用钝器分离后可见一个不透明的栓,无出血,皮损多发性,疼痛,一般7~10mm大小。通常限于跖部的承重部位
7 -
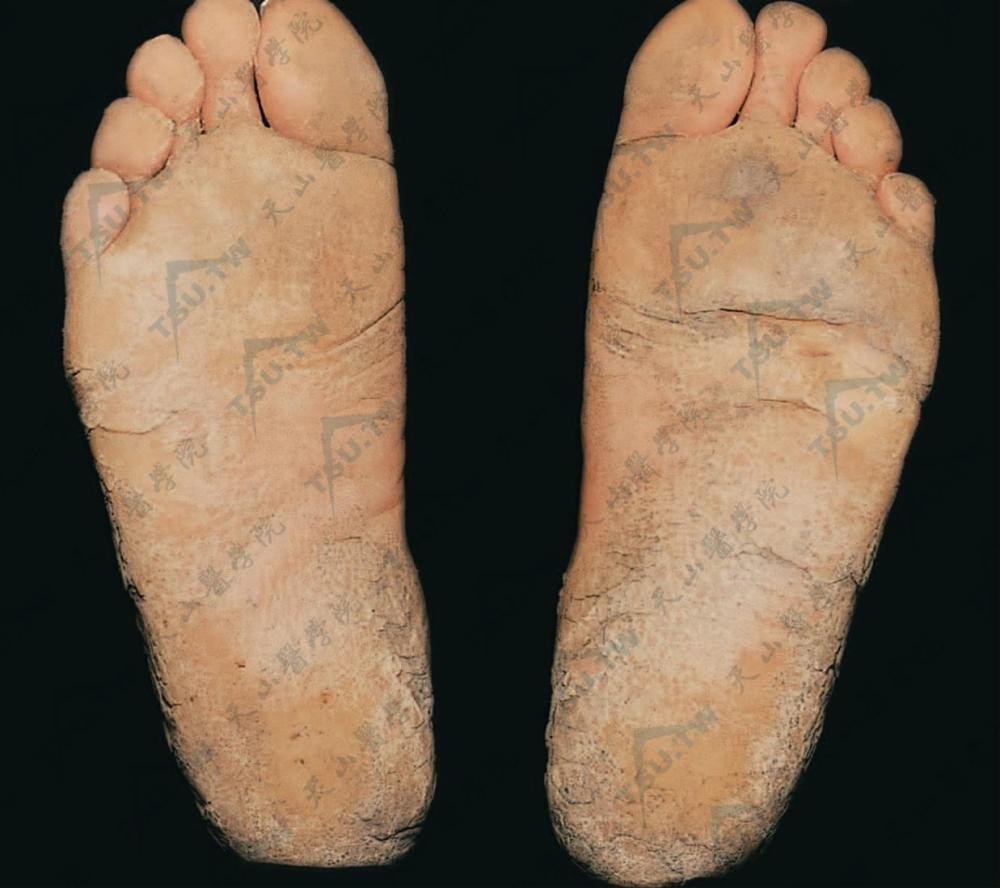
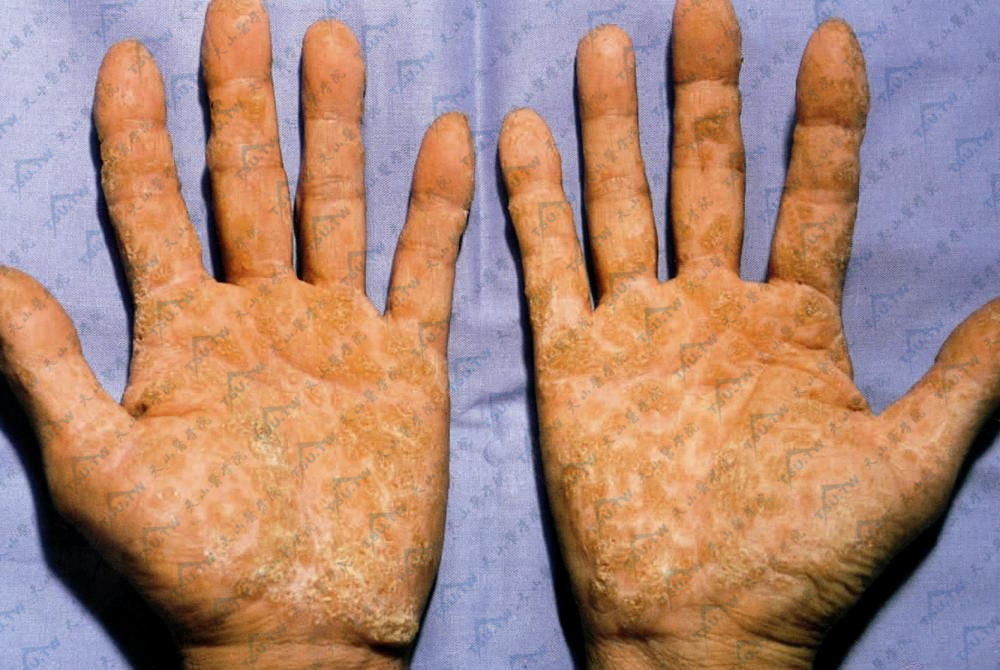
掌跖角皮症
本病又称掌跖角化病(keratosis palmaris et plantaris),这是以手掌和足跖皮肤增厚、角化过度为特点的一组慢性皮肤病。大多为先天性,常有家族史,分为显性Ⅹ连锁和隐性遗传,也包括
8 -
弥漫性掌跖角皮症
本病又称遗传性掌跖角化症(keratoderma palmo-plantare hereditaria)、对称性掌跖角质瘤(keratoma palare et plantare symmetricum)、胼胝症(tylosis)、寿斯特-乌纳综合征(Thost-U
9 -
点状掌跖角皮症
本病又称播散性角质瘤(keratoma disseminatum)、掌跖播散性角皮症(keratoderma disseminatum palmare et plantare)、丘疹性掌跖角化症(keratosis papular palmaris et plantaris
10 -
条纹状掌跖角皮症
本病又称线状角皮症、肢端角化病、布伦纳诺-富斯-西门子综合征(Brunam-Fuhs-Siemens syndrome),为一种少见的局灶性掌跖角皮症。病因及发病机制:属于常染色体显性遗传。最近的研
11 -
掌跖角皮症伴发食管癌
本病即豪威尔-埃文斯综合征(Howel-Evans syndrome)。1958年,Howel-Evans报道两个家族,其成员中患掌跖弥漫性角化过度者,有70%在以后发生了食管癌,而无掌跖角皮症的成员则以后不发生
12 -
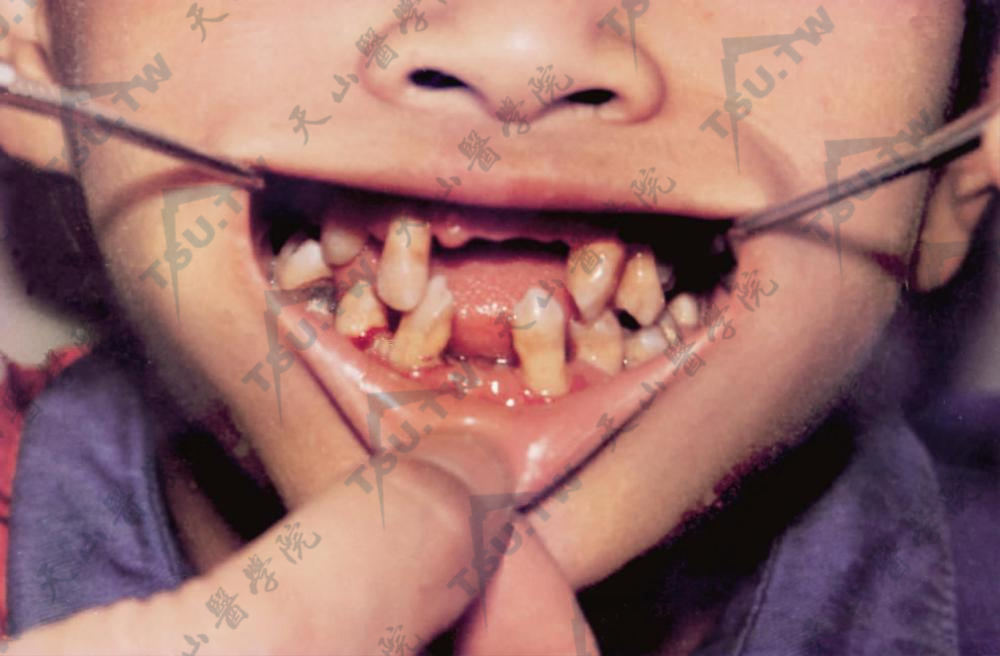
掌跖角皮症伴发牙周病
本病又名帕皮永-勒菲弗综合征(Papollin-Lèfèvre syndrome),为一种少见的常染色体隐性遗传性疾病。由编码溶酶体组织蛋白酶的基因突变引起,最近报道本病与Capthasi
13 -
进行性掌跖角皮症
本病又称格赖特尔综合征(Greither syndrome),是弥漫性非表皮松解性常染色体显性遗传型掌跖角皮症的典型变型,少见。但Itin等认为从发病史及独特的临床表现来看,本病是一种独立的
14 -
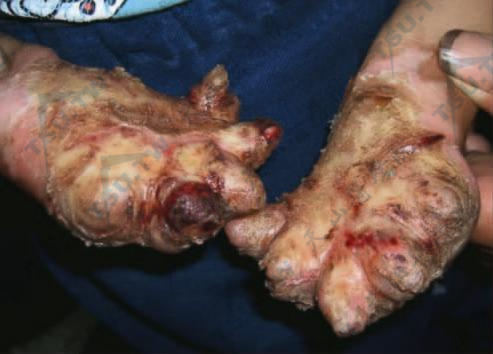
残毁性掌跖角皮症(沃温克综合征)
本病又名残毁性遗传性角质瘤(keratoma hereditaria mutilans)或沃温克综合征(Vöhwinkel syndrome),系1929年由Vöhwinkel首先描述并命名,Gibbs等强调本病的家族性发病。
15