-
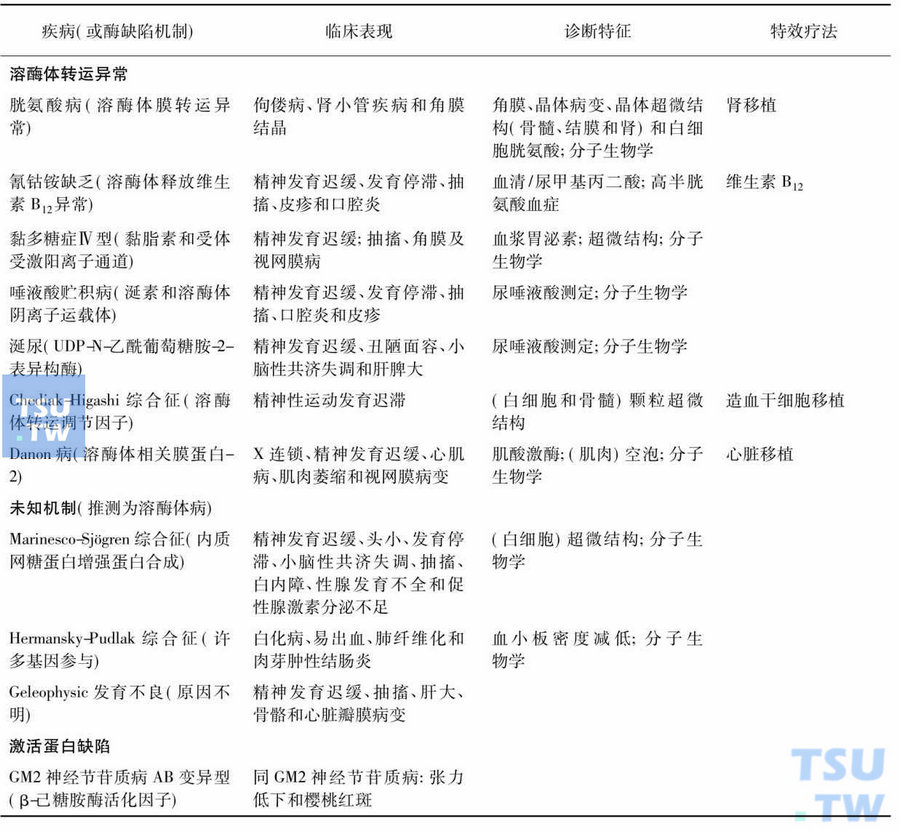
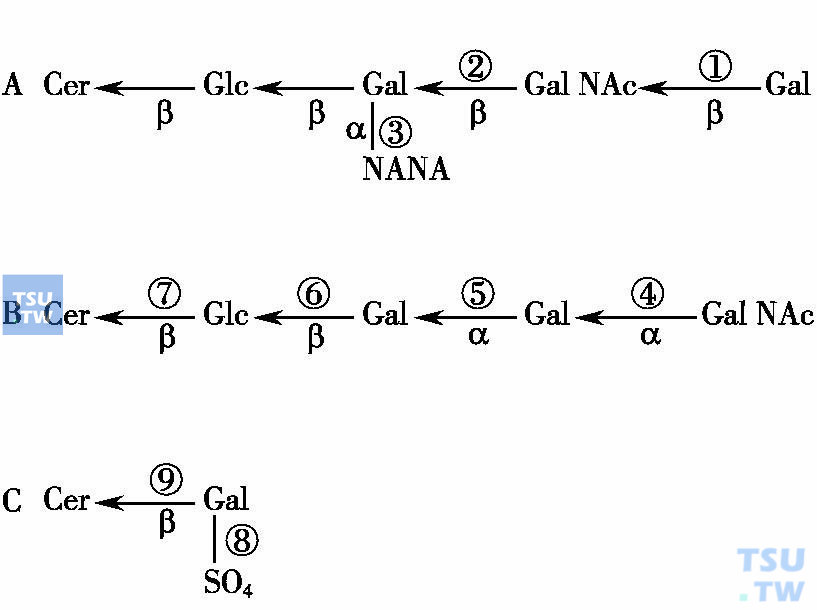
概述:神经鞘脂贮积症
由于降解鞘脂所需的溶酶体酸性水解酶缺陷或缺少了神经鞘脂激活蛋白,造成了不同的鞘脂如糖鞘脂(glycosphingolipids)、神经节苷脂(gangliosides)或鞘磷脂(sphingomyelin)在溶酶体中
2 -
戈谢病(Gaucher病)发病机制、症状表现及诊断治疗参考
戈谢病(Gaucher病)是溶酶体糖脂贮积症中较常见的一种,为常染色体隐性遗传。法国皮肤科医生Gaucher P于1882年首先报道。是因溶酶体内的酸性β-葡糖苷酶(又称葡糖脑苷脂酶,glu
3 -
尼曼-匹克病(Niemann-Pick disease,NPD)A型及B型诊治
尼曼-匹克病(Niemann-Pick disease,NPD)是一组疾病。患者除表现有肝脾大外,骨髓、脑及脏器中可见到充满脂质的泡沫样细胞,常称为尼曼-匹克细胞。NPD可分为A、B及C三型。近年来已
4