
2-噻吩乙酸
方法1 李长波,赵国诤,张洪林等.化学与生物工程,2007,24(4):22.
的合成方法路线及其结构式")
乙酰噻吩正丁胺席夫碱(3):于反应瓶中加入1.5 g甲酸,75.6 g(0.6 mol)2-乙酰噻吩(2)和61.3 g(0.84 mol)正丁胺,260 g甲苯,搅拌下中共沸脱水。减压浓缩回收甲苯和胺的混合物,得化合物(3)108.2 g,产率为89.6%,纯度90%(GC),bp 101~103.5℃/266.6 Pa(文献值102~105℃)。
N-丁基噻吩乙硫酰胺(4):于反应瓶中加入上述化合物(3)43.6 g(0.214 mol)和9.6 g(013 mol)硫和196 g(200 mL)吡啶,于100℃加热6 h。冷至40~50℃,减压蒸馏回收吡啶。向剩余物中加入160 mL甲醇,在0~5℃条件下静置4 h。过滤除去黑色不溶物,常压蒸馏回收甲醇。得42.2 g油状物(4),直接用于下步反应。
噻吩乙酸(1):于反应瓶中加入上述化合物(4)42.2 g,200 mL庚醇和32 g(0.8 mol)氢氧化钠,在135℃加热1 h。冷至室温,搅拌下加入200 mL水。分离下层水相,再用50 mL水洗涤有机相。合并水层,加入20 mL甲苯,然后在5℃用浓盐酸酸化至pH4。过滤除去不溶物,继续酸化至pH2。用二氯乙烷萃取酸化后的水溶液及其沉淀物,浓缩有机相,减压蒸馏,得到2-噻吩乙酸粗品17.3 g,产率50.9%(以2-乙酰噻吩计)。用石油醚重结晶,活性炭脱色,在0~5℃析晶。过滤,干燥,得到白色鳞片状晶体(1)15.8 g,产率46.5%(以2-乙酰噻吩计),纯度99.9%(GC),mp 63.5~65.5℃(文献值64℃)。
方法2 郭海昌,陈志荣,尹红.中国医药工业杂志,2004,35(4):201.
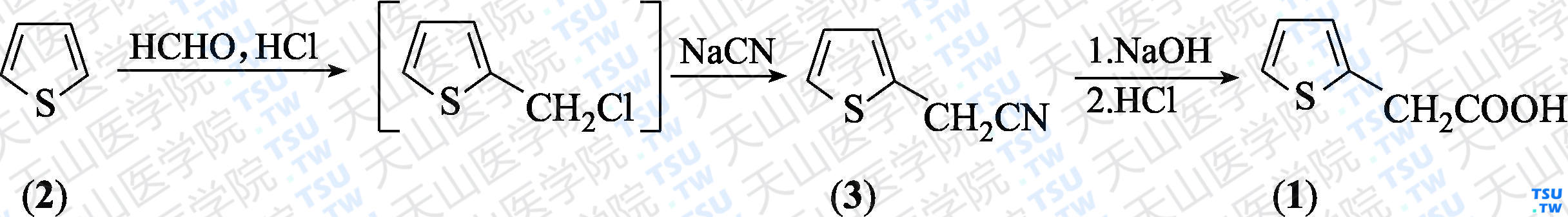
2-噻吩乙腈(3):于反应瓶中加入噻吩(2)126 g(1.5 mol)和浓盐酸129 mL(1.5 mol),于-5~0℃搅拌下40 min内滴加37%甲醛溶液121.6 g(1.5 mol),同时通入氯化氢气体,并维持饱和。加完后继续反应3 h(持续通入氯化氢)。加入冷水180 mL,分出有机层。有机相用冷水洗涤,即得淡黄色含2反应液150 mL(183 g)。于另一反应瓶中加入氰化钠72 g(1.5 mol)和水150 mL,搅拌至溶后加入如上所得的含2反应液、丙酮100 mL和相转移催化剂氯化苄基三乙基铵1.5 g,于55~65℃下搅拌反应4 h。冷后加水200 mL,水相用二氯甲烷(100 mL×3)萃取,合并有机相,用无水硫酸钠干燥。过滤,滤液常压蒸馏,回收未反应的噻吩(约18.9 g),再减压蒸馏,收集115~117℃/2.9 kPa馏分,得无色液体(3)95.1 g,收率61%(以扣除回收的噻吩计),含量大于99%(GC面积归一化法)。
2-噻吩乙酸(1):于反应瓶至加入30%氢氧化钠水溶液65 g(0.49 mol),搅拌下于95℃滴加上述化合物(3)50 g(0.41 mol),约30 min加完,再回流反应4 h。加水25 mL,降温到10℃以下,用浓盐酸调至pH 1~2,冷至5℃以下析晶。过滤,滤饼于40℃烘干,得1粗品55.9 g,收率92.3%,含量96.1%。用石油醚重结晶,活性炭脱色后于2℃以下析晶。过滤,滤饼于40℃烘干,得银白色片状固体(1)50.8 g,收率94.2%,含量99.7%(酸碱滴定法),mp 63~64℃。