-
血脂谱异常症的病因与发病机制
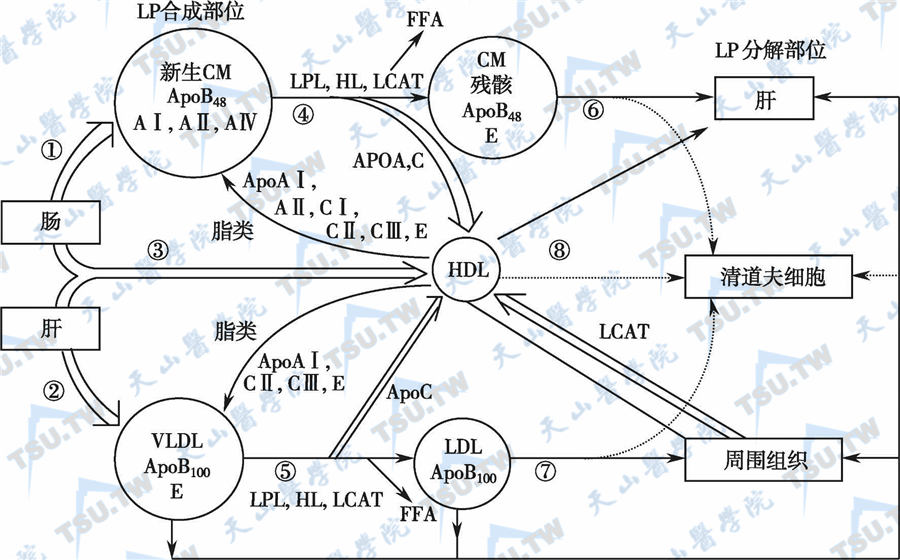
血脂谱异常症(dyslipidemia)又称为高脂血症(hyperlipidemia),是指血浆中的脂蛋白谱异常,一般特指甘油三酯和LDL-C升高伴或不伴HDL-C降低。人群中的血脂水平呈钟形正态分布,正常与异
5 -
血脂谱异常症的病理生理与临床表现
血脂谱异常症的病理生理复杂,而血脂谱异常症本身没有特殊的临床表现。肥胖、皮肤黄色瘤、动脉粥样硬化和非酒精性脂肪肝是血脂谱异常症的间接表现。血脂谱异常导致动脉粥样硬
6 -
血脂谱异常症的诊断与鉴别依据
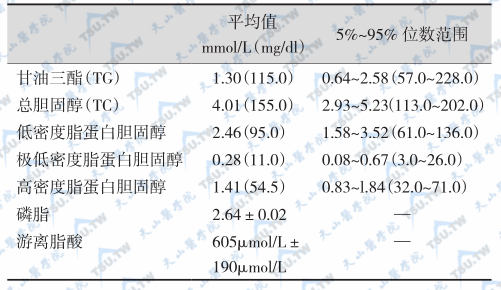
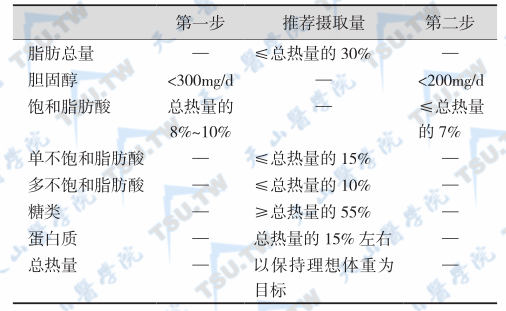
根据血脂谱异常类型与病因评估心血管病风险确立血脂谱异常症多数学者认为,血浆总胆固醇浓度大于5.2mmol/L(200mg/dl)可确定为高胆固醇血症;血浆TG浓度大于2.3mmol/L(200mg/dl)为高
7 -
家族性载脂蛋白B100缺陷症
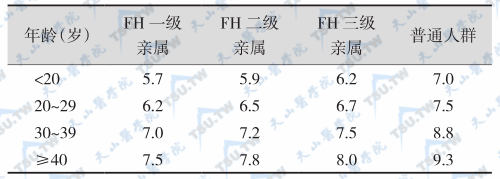
家族性载脂蛋白B100缺陷症 (familial defective apolipoprotein B-100)是一种较常见的脂质代谢性疾病。据估计,人群中家族性载脂蛋白B100缺陷症的发生率高达0.5%。载脂蛋白B100
10 -
家族性混合性高脂血症
家族性混合性高脂血症 (familial combined hyperlipidemia)呈常染色体显性遗传,与脂蛋白酯酶缺陷和胰岛素抵抗有关。表现为血浆胆固醇和甘油三酯水平升高及冠心病,但高密度脂蛋
11 -
家族性异常β脂蛋白血症
家族性异常β脂蛋白血症 (familial dysbetalipoproteinemia)又称为Ⅲ型高脂蛋白血症。ApoE常染色体显性突变患者罕见。多数属于ApoE常染色体隐性突变,多见于男性。家族性低
12 -
家族性高甘油三酯血症
家族性高甘油三酯血症(familial hypertriglyceridemia)常见,常染色体显性遗传。由于患者存在肝脏合成甘油三酯过多的代谢缺陷,使富含甘油三酯的大分子极低密度脂蛋白增多。患者
13 -
脂蛋白脂酶缺陷症
脂蛋白脂酶缺陷症(lipoprotein lipase deficiency)少见,常染色体隐性遗传。病因脂酶家族包括脂蛋白脂酶、肝脂酶和内皮细胞脂酶。在内皮细胞脂酶的作用下,内皮细胞不断分泌甘油
14 -
载脂蛋白CⅡ缺陷症
载脂蛋白CⅡ(apolipoprotein CⅡ)缺陷症是一种少见的常染色体隐性疾病,发病率低于1/100万。Apo-CⅡ是脂蛋白酯酶的激活因子,Apo-CⅡ缺陷导致功能性脂蛋白酯酶缺陷,脂蛋白脂酶不被
15