-
蛋白质-热能营养不良症的流行病学与病因
蛋白质-热能营养不良症(protein-energy malnutrition,PEM)是一种以机体组织不断消耗、免疫功能低下、器官萎缩、生长发育停滞为特征的多种营养素缺乏症,而蛋白质-能量消耗(protei
1 -
蛋白质-热能营养不良症的病理生理与临床表现
消瘦型和水肿型PEM的表现重叠临床上,常将以蛋白质缺乏为主的PEM称为水肿型PEM,而将能量缺乏为主者列为消瘦型PEM,两者兼有者则为混合型PEM。但事实上,消瘦型和水肿型PEM的病因、
2 -
蛋白质-热能营养不良症的诊断与鉴别依据
目前尚无诊断PEM的统一标准。由于病程和临床类型不同,有时诊断比较困难。临床上应避免蛋白质-热能营养不良症(PEM)诊断不足和诊断过度的两种倾向。PEM诊断不足可能仍然是一种普
3 -
蛋白质-热能营养不良症的治疗
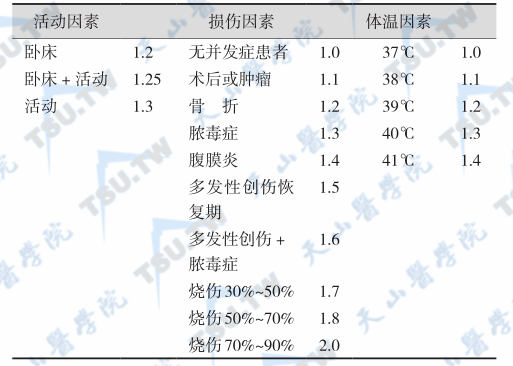
在多数情况下,蛋白质-热能营养不良症(PEM)患者病情较危重,故应尽早采取处理措施,其主要目的是: 立即改善威胁生命的PEM指标; 有步骤地恢复和补充营养物质; 确保机体在营养复原中预防
4 -
系统性淀粉样蛋白变性的分型和病理
系统性淀粉样蛋白变性(systemic amyloidosis)和Alzheimer病以及其他一些疾病的病因与致病性蛋白聚合物(pathogenic protein aggregates)不适当沉积有关。系统性淀粉样蛋白变性是
5 -
系统性淀粉样蛋白变性的临床表现与诊断
系统性淀粉样变是指在全身各种组织中和器官中均有淀粉样蛋白沉积,但有些患者只在局部沉积,其中有些患者可能是系统性淀粉样变的早期阶段,以后再发展到其他组织或脏器的淀粉样蛋
6 -
系统性淀粉样蛋白变性的治疗
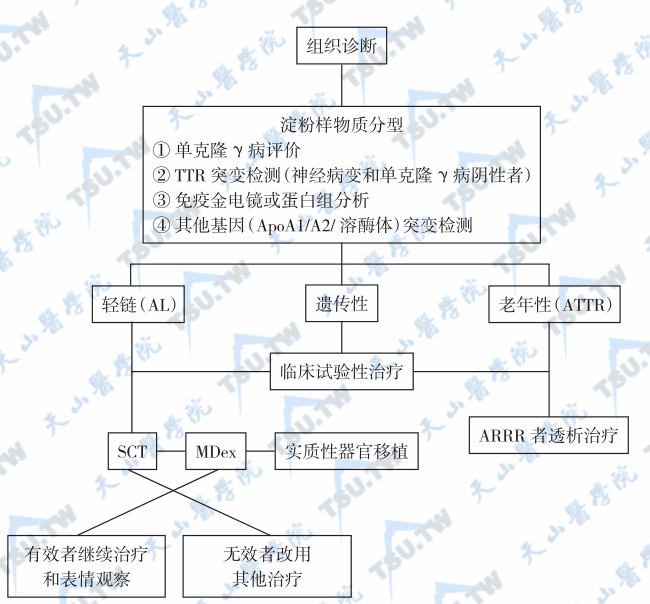
系统性淀粉样变目前无根治方法,可根据不同类型采取不同的治疗方法[24,25]。多种药物联合治疗AL型淀粉样变经典的治疗方法为联合应用美法仑(melphalan,左旋苯丙酸氮芥)和泼尼松。前
7 -
枫糖尿症及其分型
枫糖尿症(maple syrup urine disease)是一种遗传性支链氨基酸(branched chain amino acids,BCAA)代谢障碍的疾病,由于细胞线粒体基质内支链α酮酸脱氢酶(branched-chain alpha
8 -
枫糖尿症的病因与发病机制
本病属遗传性疾病,呈常染色体隐性遗传。遗传缺陷在编码线粒体中存在着BCKD多酶复合体中的E1、E2和E33几个亚基的基因突变。支链氨基酸代谢障碍引起枫糖尿症BCKD多酶复合体的
9 -
枫糖尿症的临床表现与诊断依据
本病的临床表现与BCKD复合体活性降低的程度有关,而BCKD复合体活性是由E1、E2和E3基因突变所决定。枫糖臭味最具诊断意义从典型表现到只有轻微症状,典型型出生后即可发病,患者的
10 -
枫糖尿症的治疗
本病虽不能根治,但及时正确治疗可使患儿存活,症状可得到改善。治疗原则包括: 抑制内源性蛋白质分解代谢; 保持蛋白质合成正常进行; 供给必需氨基酸(包括亮氨酸、异亮氨酸、缬氨酸),
11 -
苯丙酮尿症(PKU)分型与发病机制
苯丙氨酸(phenylalanine,PKU)是一种必需氨基酸,食入的苯丙氨酸如未用于合成蛋白质,正常是通过酪氨酸途径降解,缺陷苯丙氨酸羟化酶或者它的辅酶四氢生物喋啶,会导致苯丙氨酸在体液中
12 -
苯丙酮尿症的临床表现与辅助检查
苯丙酮尿症(PKU)是一种遗传性疾病,故新生儿期即有高苯丙氨酸血症,由于未进食,血苯丙氨酸及其有害的代谢产物浓度不高,故出生时无临床表现。如果对新生儿未作苯丙酮尿症筛选,随着喂养时间延长,血中苯丙氨酸及其代谢产物逐渐升高,临床症状才逐渐表现出来。
13 -
苯丙酮尿症(PKU)诊断与鉴别依据
应强调早期诊断,以便得到早期治疗,避免智力发育障碍。要得到早期诊断必须在新生儿中进行苯丙酮尿症的筛查。从生长发育障碍伴棕色毛发及尿霉臭病例中筛查PKU下列表现提示苯丙
14 -
苯丙酮尿症的治疗与预防
PKU是遗传性疾病,PAH基因突变是无法根治的,但可避免高苯丙氨酸血症所带来的严重后果。治疗方法主要是限制饮食中苯丙氨酸摄入量。饮食治疗饮食治疗原则是既要限制饮食中苯丙氨
15