
表皮角质形成细胞通过细胞膜上的桥粒与半桥粒为角蛋白纤维网提供附着点,桥粒还将角质形成细胞相互连接,半桥粒则将基底细胞固定在基底膜带上,后者通过由Ⅶ型胶原组成的锚纤维将表皮与真皮连接在一起,从而保证表皮的完整与功能。同时,角质形成细胞连接结构成分也是细胞间信息传递的枢纽。不难理解这些连接结构成分先天缺陷可以导致遗传性皮肤疾患。
桥粒原发性缺损相关皮肤疾病
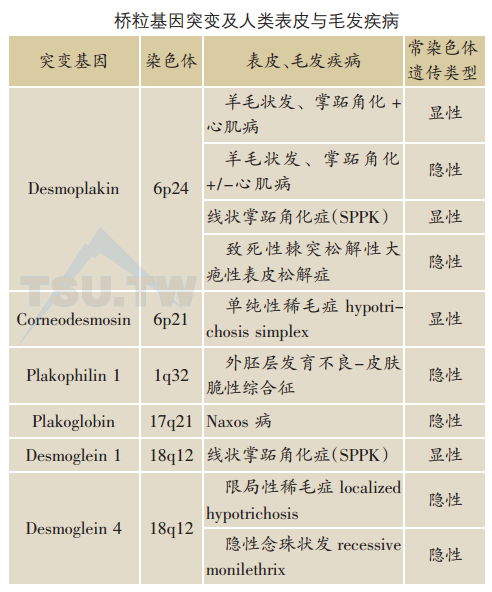
桥粒是表皮细胞及心肌细胞间的主要连接结构,过去十多年来已发现8种人类基因变异所致皮肤疾病与桥粒成分有关,涉及桥粒介导的细胞间黏附异常(下表)。
桥粒斑蛋白
桥粒斑蛋白(desmoplakin,DP)基因突变与一组合并有皮肤、毛发及心肌病变的异常DP基因突变与先天性遗传性异常的关系源于对一组伴有线状掌跖角化症(SPPK)常染色体显性遗传家谱的研究。异常基因图定位于染色体6p24,其中包括DP基因位点。进一步DNA序列筛选在DP基因上检测出杂合子无义突变p.Q331X。这种突变使肽链合成过早终止从而导致单倍体功能不全,50%正常水平的DP蛋白足以保障非掌跖部位正常表皮的结构及功能,但不足以抵抗掌跖部位的外力损伤。自1999年以来,共有17种不同的DP基因突变被发现。DP基因突变的表现型可以多样,可以是常染色体显性,也可以是隐性。临床主要表现为皮肤、毛发异常,或伴心脏异常,包括掌跖角化、羊毛状发及心室扩张心肌病。皮损表现为线状掌跖角化症,DP突变蛋白严重缺损可引起致死性棘突松解性大疱性表皮松解症,表现为进行性泛发性表皮松解,伴有弥漫性脱发、甲缺失及牙齿发育障碍。
斑菲素蛋白1
斑菲素蛋白1 (plakophilin 1,PKPI)基因突变与外胚层发育不良一皮肤脆性增加综合征(ectodermal dysplasia-skin fragility syndrome) PKPI(又称为带6蛋白)基因位于染色体1q32。PKP1蛋白属Armadillo家族,约80.5 kD。其基因突变是最早发现的人类桥粒成分原发性缺损变异。McGrath等于1 997年描述了一例患儿皮肤脆弱伴炎症(皮肤糜烂水疱、皲裂、鳞屑、结痂、掌跖角化)合并外胚层发育不良(身材矮小、毛发稀疏、少汗及散光)。表皮细胞分析发现患儿遗传了复合型杂合子PKP1基因失功能突变,父系等位基因910碱基位点C→T点突变导致谷氨酰胺基团被终止码取代(Q304X),母系等位基因在1 132碱基位点插入一段28个碱基对内部重复序列(1132ins28)引起移框突变,并导致终止密码子提前产生而使肽链合成过早终止。自此,又有8例带有不同PKP1基因突变的临床类似病例报道。
桥粒斑珠蛋白
桥粒斑珠蛋白(plakoglobin,PG)基因突变与Naxos病Naxos病是目前所发现的仅有的PG基因突变所致疾病。Naxos病是一种伴有心肌病、皮肤及毛发异常的常染色体隐性遗传性疾病,表现为非表皮松解性掌跖角化症(NEPPK)、羊毛状发和致心律失常性右室心肌病(arrhythmogenic right ventricular cardiomyopathy,ARVC)。患者cDNA分析发现在PG基因第2157碱基位点上有TG2个碱基对缺失(c.2157deITG)。Naxos病的命名来源于地中海Naxos岛,岛上每1 000人中就有1人发病。所有的患者均为同样的c2157deITG纯合子突变,导致PG蛋白C末端移框突变及终止码提前产生。
副肿瘤性天疱疮(paraneoplastic pemphigus)是一种新近描述的皮肤-黏膜水疱性病变,发生在肿瘤患者,患者体内可检测到抗桥斑蛋白Ⅰ/Ⅱ自身抗体。可能是由于细胞损伤,抗原释放所致。
corneodesmosin
corneodesmosin(CDSN)突变与单纯性稀毛症CDSN除在表皮角质层外,还在毛囊内根鞘表达。自2003年以来研究者在三个单纯性稀毛症家系的CDSN基因上分别发现三种基因无义突变,即p.Q215X、p.Q200X或p.Y239X。组织学上,毛囊内根鞘失去黏附性,毛囊周围及真皮浅层有异常蛋白降解的CDSN聚集。研究提示CDSN基因突变导致蛋白折叠异常是常染色体显性单纯性稀毛症病因。
桥粒芯糖蛋白1
桥粒芯糖蛋白1(Dsg1)基因突变与线状掌跖角化症除了遗传性桥粒致密斑蛋白基因突变外,钙黏素家族蛋白细胞外结构变异也可导致病变。Dsg1基因突变所致掌跖角化症首先由Rickman于1999年描述,与Armstrong所描述的DP变异单倍体功能不全的表现型相似。Dsg1基因突变为拼接接受位点突变,转录时跳过外显子2~4而产生仅有25个氨基酸的多肽,从而导致Dsg1单倍体功能不全而致病。临床主要表现为掌跖角化,典型为条带状,也可为局灶型。Dsg1基因突变皮损还常伴随基底层上桥粒减少或减小,并伴有K5、K14、K10及K16表达降低。
桥粒芯糖蛋白4
桥粒芯糖蛋白4( Dsg4)基因突变与毛发异常Dsg4基因突变是限局型常染色体隐性稀毛症的病因。患者表现为毛发稀少,涉及头皮、胸及四肢,而腋毛、阴毛及眼睫毛则不受累及。于2003年首先发现的Dsg4基因突变为EX5-8del,导致整个第2细胞外功能基团及部分第1及第3细胞外功能基团缺失。第2例Dsg4基因错义突变p.A129S于2005年报道,这种氨基酸取代突变位于—个重要的细胞黏附功能区。自2006年以来,又相继有几例Dsg4基因错义突变、拼接处突变、蛋白翻译框架移位以及失义突变的报道,5种基因突变位于Dsg4细胞外功能基团,1种突变位于跨膜功能基团。但其表现型为常染色体隐性念珠状发,不同于毛发角蛋白基因KHB1、KHB3和KHB6基因突变所致的常染色体显性遗传念珠状发。有时常染色体隐性稀毛症及常染色体隐性念珠状发表现可以重叠。
另外,落叶性天疱疮(PF)患者体内有抗Dsg1的自身抗体,而寻常性天疱疮(PV)患者体内出现抗Dsg3的自身抗体,从而引起细胞失去彼此间的相互黏附,导致表皮内水疱。水疱的位置与这两种桥粒芯蛋白(Dsg1及Dsg3)在表皮内的分布相对应。
半桥粒以及基底膜相关性疾病
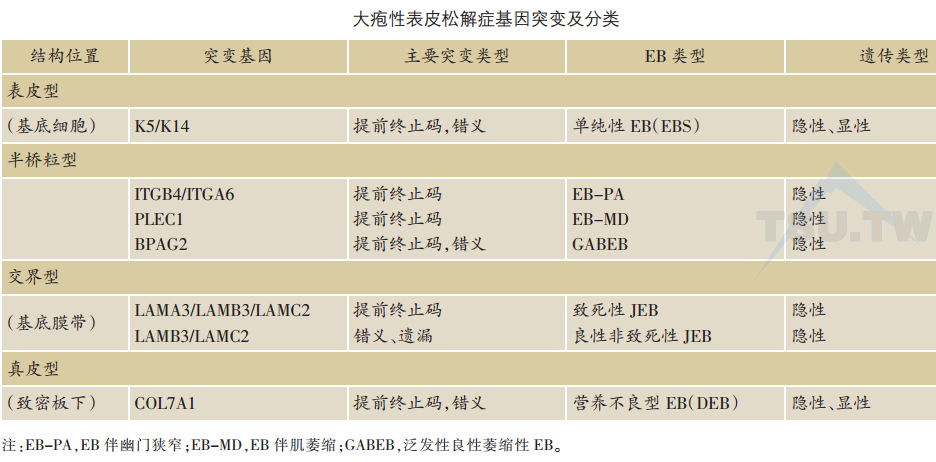
近年来由于对表皮、真皮交界结构特征的理解,主要是通过对这些结构成分基因的分子克隆,在多种皮肤遗传性水疱性疾病上的研究取得了明显的进展。表皮、真皮连接结构成分基因的突变或表达异常是临床上多种遗传性水疱性皮肤病的发病基础。其中,大疱性表皮松解症(EB)是这组疾病的代表。根据裂隙所在的位置,EB大致可以分为三种:表皮型单纯性EB,组织分离出现在基底细胞层,主要是由于角蛋白基因K5及K14的突变所致,本章第一节已叙及。交界型EB(JEB),裂隙见于基底膜带,主要位于透明板,突变在于合成锚丝蛋白板层素5三条不同肽链的基因。近年来,半桥粒本身结构成分基因的突变,如BPAG2、α6β4整合素及plectin,被发现与某些JEB有关,近来趋向于单独归于半桥粒型EB。在这些病变中,裂隙形成在半桥粒水平,见于基底细胞与基底膜带透明板交界处,临床上常伴有其他系统症状。在真皮型(营养不良型)EB,组织分离出现在基底膜带致密板下方,是连接基底膜致密板与其下方真皮的锚纤维组成蛋白Ⅶ型胶原基因突变所致(下表)。
交界型EB
交界型EB(JEB)板层素5基因突变JEB为隐性遗传,分为致死性( Herlitz)及良性非致死性(非Herlitz)两种。参与合成板层素5的α3、β3及γ2三条肽链的三种基因LamA3、LamB3以及LamC2在致死性JEB中均分别发现有突变,而LamB3和LamC2两种基因的突变还见于非致死性JEB。最常见的是LamB3基因突变。常见的为单一碱基突变C→T,从而引起精氨酸(CGA)被终止码(TGA)所取代。常见突变类型为R42X、Q243X、R635X以及957ins77。在致死性JEB中,基因突变均引起提前产生的终止码(PTC/PTC)。在某些非致死性JEB中,突变还包括错义突变以及外显子遗漏突变。
半桥粒型EB
半桥粒基因突变而致的EB根据其临床伴随的症状可分为三组,包括伴有肌萎缩(EB-MD)、幽门狭窄(EB-PA)或泛发性良性萎缩(generalised atrophic berugn EB,GABEB)。
α6β4整合素基因突变:α6β4整合素基因突变被发现与EB伴幽门闭锁(EB-PA)有直接的联系。常见为β4基因(ITGB4)突变(大于90%),α4基因(ITGA4)突变较少。患者表皮内β4表达大大降低。cDNA序列分析发现,ITGB4或ITGA4基因多为提前终止码无义突变、插入或遗漏突变、错义突变或拼接位点突变。有一例患者母系同源染色体β4基因上有单个T碱基插入,导致5’端切割位点丢失,以及β4细胞内功能区第2个纤维连接素样重复序列中51个核苷酸丢失(相当于碱基位置第3750~3801),称为3801+2insT突变。患者父系同源染色体β4基因上,在第1150位点有单个G的丢失(1150△G),导致产生提前终止码,以致β4肽链截断在第409个氨基酸位置。
BPAG1与BPAG2基因突变: 分子量为230 kD的BPAG1及分子量为180kD的BPAG2最初被发现是大疱性类天疱疮的自身抗原,目前已知BPAG2自身抗原还见于妊娠疱疹及良性黏膜类天疱疮。最近的研究还发现,这两种半桥粒组成成分可以通过两种不同的病理机制致病:一是通过BPAG1以及BPAG2自身抗体,另外一种是通过基因突变。
近来发现一些伴有泛发性萎缩性良性EB(GABEB)的病变基础在于BPAG2( COL17A1)基因突变。突变包括提前终止码(PTC)无义突变、插入或遗漏突变、错义突变或拼接位点突变。在一例病变中,基因突变表现为两种无义复合突变。父源性基因上为单个碱基C→T突变,导致第1226个氨基酸基团由精氨酸(CGA)被终止符(TGA)所取代,称为R1226X。而母源性基因上在第4150个碱基位点处有单个碱基G插入(4 150+G),引起编码框移位。两种突变均导致提前出现的终止码(PTC/PTC)。
新近有报道BPAG1去基因鼠表现类似于人类的JEB伴肌无力。尽管这些去基因鼠皮肤半桥粒大致正常,但半桥粒缺乏内斑块,并完全失去与角蛋白中间丝的连接,水疱位于半桥粒的正上方。由BPAG1去基因鼠表现的症状与人JEB伴肌营养不良十分相似,提示BPAG1很可能在这种病变的病理机制中起着重要的作用。对这种疾病基因缺损的鉴别将有助于对皮肤与神经系统的关系,以及细胞骨架及其相关蛋白在神经变性损害中作用的认识。
网蛋白基因突变:研究发现网蛋白基因(PLEC1)突变与一种伴有肌萎缩的JEB发病有关(EB-MD)。EB-MD是一种比较少见的常染色体隐性遗传,临床上患者表现为新生儿水疱、较晚发生的进行性肌萎缩以及指甲、牙齿异常,也有报道有严重的黏膜受累。最早用抗网蛋白抗体研究发现患者角质形成细胞中网蛋白完全消失,继后进一步研究显示在一些家系中有PLEC1基因突变。网蛋白基因突变可为无义突变、插入或遗漏突变、拼接位点突变,多数突变导致提前终止码产生。近来也有报道网蛋白基因突变见于EB-PA。
Ogna型EBS是另一少见的网蛋白基因突变所致常染色体显性遗传,在网蛋白基因棒状功能基团内可检出一种特异的错义突变(R2110W).裂隙位于表皮基底细胞。临床上患者表现为皮肤出血性水疱,但不同于EB-MD的是,患者没有肌萎缩,肌肉活检网蛋白染色正常。
真皮型(营养不良型)大疱性表皮松解症
简称DEB,可为显性遗传,也可为隐性遗传。DEB在超微结构上表现为基底膜带致密板下锚纤维形态异常,数量减少或完全消失。免疫病理上,抗Ⅶ型胶原抗体免疫荧光染色常有异常。在轻型显性DEB,免疫染色常接近正常。在轻型隐性DEB,免疫荧光染色减弱。而在重型泛发性隐性遗传性Hallopean-Siemens DEB (DEB-HS),免疫荧光则完全阴性。分子遗传学上,基因连锁研究将DEB基因遗传缺损定位于染色体3p21COL7A1位点,即Ⅶ型胶原基因所在位点,进一步证实DEB是一种Ⅶ型胶原疾病。
近来对Ⅶ型胶原整个基因的克隆成功以及cDNA序列的阐明,为分析DEB各种突变提供了可能性。1992-2007年,在COL7A1基因上已发现324种致病性突变,多为无义突变、错义突变、遗失或插入以及拼接位点突变。DNA序列分析表明,DEB临床损害的严重性与基因突变类型有对应性。报道最多的是重型毁损性DEB-HS,至今为止已有多例DEB-HS的两种基因突变均已被发现。所有的突变均引起两个等位基因上终止码提前出现(PTC/PTC),进而导致锚纤维的完全消失。在轻型隐性非Hallopean-SiemensDEB( DEB-nHS),突变多为一个基因上为PTC,而另一个基因上为错义突变或甘氨酸被替换。第二种突变常不影响合成全长Ⅶ型胶原分子,这种胶原蛋白分子可能组装成锚纤维,但不能被二硫键所稳固,从而临床上损害较轻。在显性DEB,所检出的基因突变均为在三螺旋胶原样编码区单个甘氨酸被替换。这种氨基酸替代突变的影响表现在翻译后水平,可以影响三螺旋结构的稳定,阻碍蛋白分子的分泌并促使蛋白降解。由于Ⅶ型胶原是同种三聚体,形成正常三螺旋结构分子的几率为1/8,因此常可见部分正常锚纤维以及相对较轻的临床症状。
