
皮肤疾病时表皮的变化
表皮细胞动力学的变化
在人的一生中,细胞不停分裂以维持分化的组织并代替死亡的细胞,但是在神经细胞、骨骼肌细胞不存在细胞分裂。许多组织包括皮肤和黏膜(复层鳞状上皮)以及胃肠道(单层上皮)中,一小群干细胞快速而持续地更新为分化细胞。传统上认为表皮是一复层鳞状上皮,依靠基底层细胞不断分裂而维持其完整性。分化的细胞逐渐由棘细胞层迁移至角质层并失去细胞核,无核的角质层细胞保护有活力的角质形成细胞并从皮肤表面不断脱落。基底层生成的细胞需与角质层丢失的细胞比率匹配以维持正常的表皮厚度,但在病理状态下这种比率的平衡被打破。
表皮干细胞:干细胞具有无限自我更新的能力,产生子代细胞并经历终末分化。但是,并非所有具有分裂能力的基底细胞都是干细胞。经历终末分化的干细胞子细胞必然要首先增殖,分裂数次(约5~6次有丝分裂),这类细胞称为短暂扩增细胞(transient amplifying cells),又称角质形成细胞分化池。增殖细胞的扩展增加了来自每个原始干细胞的终末分化角质形成细胞的数量,因此表皮干细胞虽然具有巨大的增殖能力,但是实际上并不经常处于分裂状态。
表皮干细胞位于表皮基底层和毛囊间上皮,特别是毛囊的毛球部位。表皮干细胞在形态上类似角质形成细胞,但具有特殊的分子生物学标记。
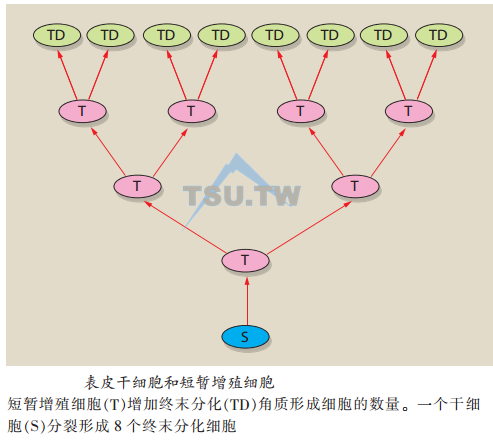
毛球部位的干细胞具有迁移的能力,在毛囊再生时迁移至基底部。毛囊干细胞还可分化为多种细胞系,如外毛根鞘、内毛根鞘、毛干、皮脂腺细胞及滤泡间上皮细胞。具体的终末分化方向由一些局部微环境因素控制,包括Shh蛋白(sonic hedgehog)、Wnt和BMP(bone morphogenetic protein,骨形态发生蛋白)途径,它们在胚胎和出生后毛囊的发育过程中发挥重要的作用。
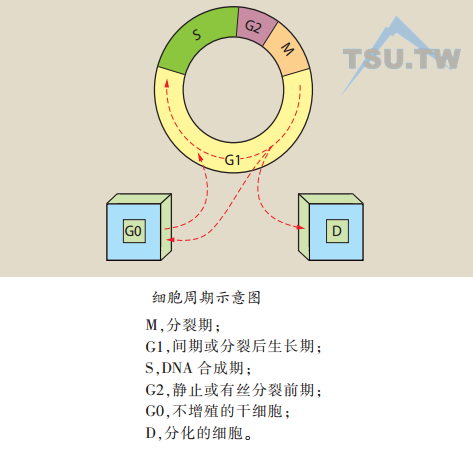
表皮细胞动力学:表皮细胞动力学是细胞生长分化和细胞死亡之间的复杂平衡过程。分化的细胞可能不具有增殖能力但具有广泛的代谢活性,在细胞数目不增加的情况下增加组织的体积。在皮肤生物学的研究中有几个重要的细胞动力学概念。一个重要的概念就是更新时间(turnover time),是整个细胞群代替自身的时间,又称再生时间或更替时间(regeneration time或replacement time),取决于单个细胞分裂的时间、细胞周期以及基底细胞的生长比。
- 细胞周期或有丝分裂间期时间(intermitotic time,Tc):指两次连续的有丝分裂之间的间期。细胞周期根据分裂过程又可分为下列各期:分裂期(M)、分裂后生长期(G1)、DNA合成期(S)和分裂前生长期(G2),所有增殖的细胞按照周期进行。某些基底细胞停留在静止期(G0)期,也可在多种刺激因素作用下进入细胞周期继续增殖。细胞丢失平衡,包括细胞再生、凋亡、死亡、脱屑,决定了细胞群增加或减少的比率。正常表皮细胞周期时间从50小时(流式细胞计数)到457小时(更新时间)。
- 生长比:指某一时间具有增殖能力的基底细胞的比例,在正常小鼠细胞生长比约为60%。细胞周期缩短和高生长比导致高增值率。有丝分裂指数指在某时间点基底细胞或活细胞处于有丝分裂的比例。
- 表皮更新时间(epidermal turnover time)或表皮通过时间(transit time):指细胞从基底层到达皮肤表面的时间,包括表皮细胞由基底层增殖、分化过渡到棘细胞层以及进一步分化为无活性的角质层的时间。可以通过注射放射性标记或荧光染料标记估计表皮通过时间,正常皮肤的表皮通过时间为52~75天,但在银屑病等这一时间大大缩短。
银屑病表皮细胞周期维持在50小时左右(体外培养为22~24小时),生长比为100%.表皮更新时间大大缩短,为3~5天。细胞分化改变,表现为颗粒层消失和角化不全。当然表皮生长加速并非银屑病所特有,亦可见于毛发红糠疹、苔藓样湿疹、脂溢性皮炎等。
生长刺激信号
- 表皮生长因子(EGF):人EGF是一分子量为6 kD的多肽,含53个氨基酸。EGF在皮肤组织中有多种靶细胞,包括表皮角质形成细胞、真皮成纤维细胞和皮肤血管的内皮细胞等,对细胞外基质和皮肤微环境都具有重要的调节作用。EGF是一种强效细胞增殖因子,能够加速正常皮肤表皮更新。EGF促进表皮细胞增殖必须先与基底细胞膜上特异的受体EGFR-1(分子量170 kD)结合。EGF受体是一种跨膜蛋白质,与EGF有特异的亲和性。EGF受体分子具有酪氨酸激酶活性,称为酪氨酸激酶受体。当EGF与EGFR结合后,酪氨酸激酶被激活,受体自身发生磷酸化而活化,启动信号传递过程。EGFR在正常表皮分布于基底层,而在银屑病则分布于表皮的全层。
- 转化生长因子α(TGF-α):为单链26 kD的多肽,由表皮角质形成细胞合成,与EGFR结合以自分泌的方式促进角质形成细胞增殖。TGF-α能促进其自身及EGFR-1的产生及银屑病表皮基底细胞上层TGF-α过表达。
- 双向调节因子(amphiregulin):与EGFR结合,亦以自分泌的方式作用于角质形成细胞,促进其增殖。外源性EGF和TGF-α能促进其表达。双向调节因子在过度增殖性皮肤病和鳞癌中表达增加。
- 血管内皮生长因子(VEGF):血管内皮生长因子通过血管内皮细胞特异性受体[VEGF受体(VEGF receptors,VEGFR)]介导血管内皮细胞的增殖、微血管增生及增强血管通透性。VEGF及其受体在与血管增生有关的肿瘤、缺血性疾病中已经展开一些中和性抗体、反义寡核菅酸、特异性受体阻滞剂等方面的实验性治疗。由于角质形成细胞是VEGF的重要分泌细胞,而且真皮层微血管分布广泛,一些病理情况下微血管增生显著,因此,VEGF及其受体在皮肤病中的作用越来越受到重视。在人正常表皮中,VEGF受体,包括VEGFR-1、VEGFR-2、VEGFR-3及NRP-1、NRP-2均有表达。VEGF与VEGFR结合,可以促进角质形成细胞的增殖和迁移。
- 其他:白介素IL-1、IL-6、粒单集落刺激因子(GM-CSF)、角质形成细胞生长因子(KGF)、TNFα等亦能促进细胞快速增殖。
表皮细胞分化功能的变化机制角
质形成细胞离开基底层后,它就开始了复杂的终末分化过程,最后形成角质层并脱落。在此过程中,细胞生物化学、形态学和细胞的结构均发生显著的改变,至角质细胞(鳞屑)已无细胞核与细胞器,其中65%为不溶于水、富含半胱氨酸的由二硫键联结起来的蛋白——角蛋白(keratin)。在角质层中,角蛋白丝在丝聚合蛋白作用下聚集成束状,同时由内披蛋白形成角质化包膜。此外,还可表达出细胞间脂质及一些膜糖蛋白,如生长因子受体、整合素及血型抗原等。
角蛋白:角蛋白是一种螺旋蛋白,根据分子量的不同分为30余种。根据分子量和等电点不同,角蛋白可分为酸性和碱性两个亚族或称为Ⅰ型和Ⅱ型。Ⅰ型角蛋白等电点偏酸,相对分子质量较低,基因定位于染色体17q,包括K9-K20和4种毛发角蛋白Ha1~Ha4。Ⅱ型角蛋白等电点偏碱,相对分子质量较高,基因定位于染色体12q,包括KI~K8和4种毛发角蛋白Hb1~Hb4。上皮细胞中的角蛋白按一定规律构成中间丝,其组合方式即该种上皮的CK组型,按CK组型不同可把上皮细胞分为单层上皮、复层扁平上皮和其他上皮(包括移行上皮、间皮、某些腺上皮)三大类。
在组装成角蛋白丝时,Ⅰ型角蛋白和Ⅱ型角蛋白单体的杆状区平行相联,形成长44~46 nm的异源二聚体,两个二聚体再以反平行方式侧面相交联而形成四聚体,最后以四聚体为基础形成宽约10 nm的角蛋白丝。角蛋白在皮肤中多成对存在,并依部位不同而不同,基底细胞产生K5和K14,基底层以上的细胞则可合成K1和K10,而且K1和K10共表达是表皮分化的特征之一。在外毛根鞘及甲床等处则表达出K6和K16,这种优先表达的特性主要是为了适应不同类型上皮的需要。
角质化包膜:角质化包膜位于角质细胞膜内,厚10-20 nm,约占角质层干重的10%,由于其含有大量二硫键及δ-(γ-谷氨酰)-赖氨酸异肽,故难溶于水。内披蛋白是角质化包膜的重要前体蛋白,分子量95kD,一般在棘层上部合成,位于胞质周边,可由转谷氨酰胺酶催化形成谷氨酰-赖氨酸交联,转谷氨酰胺酶在催化过程中需钙和巯基,所以角质形成细胞内钙浓度是调节角质化包膜形成的重要因素,而且转谷氨酰胺酶被认为是表皮终末分化的标志。
兜甲蛋白是最近才发现的一种与角质化包膜形成有关的蛋白,分子量为60 kD,富含甘氨酸、丝氨酸,结构中有脂族氨基酸(甘氨酸、丝氨酸、半胱氨酸)所组成的串联重复片段并交替排列成杆状。兜甲蛋白主要在颗粒层产生,位于角质化包膜内侧,当其移至角质细胞周边部位时,可使角质化包膜与角蛋白丝相连。
透明角质颗粒蛋白:透明角质颗粒的主要成分是一种无定形富含磷酸的物质,此蛋白富含组氨酸,带正电荷,在体外可促使角蛋白组装成角蛋白丝,故被命名为丝聚合蛋白,其前体物质即为丝聚合蛋白原,在合成后即被高度磷酸化,并贮存于透明角质颗粒中,在表皮分化的最后阶段,丝聚合蛋白原转变为丝聚合蛋白,磷酸则被去除,此过程若出现异常,可引起鱼鳞病。人丝聚合蛋白原基因被定位于第1号染色体。维生素A衍生物对丝聚合蛋白有明显的抑制效应。
表皮分化的调节:几种信号传导途径如hedgehog、Wnt和转化生长因子TGF-β均参与皮肤的发育过程,而且这些途径在上皮分化、增殖和肿瘤发生的过程中表现为进化保守性。这些途径激活方式各异而产生不同的转录因子。在角质形成细胞分化的过程中,3个主要的转录因子AP1、AP2和Sp1具有关联性。体外研究也发现一些关联转录因子如碱蛋白(basonuclin)、C/EBP、Oct6和Oct11、ESE2 (ELF5)、Klf4以及维A酸受体。
核转录因子NF-κB在表皮的终末分化中亦发挥重要的作用。IKKα和IKKγ磷酸化后破坏IκB( NF-κB的胞质抑制剂)。将IKKα靶向失活,则导致终末分化不能进行,即使NF-κB处于正常水平。可能某个依赖IKKα活性尚不知的分泌因子导致分化缺陷。但是,如果IKKβ失活,则NF-κB活性发生异常。IKKβ常促进NF-κB的活性。综合以上数据可以看出,NF-κB失活导致表皮生长和分化失平衡。
维A酸亦能在mRNA水平抑制终末分化,其效应发挥通过维A酸受体RARs和RXRs。RARs缺陷的转基因鼠表皮屏障功能缺陷,抑制表皮的分化。
表皮细胞黏附能力变化的机制
表皮角质形成细胞黏附能力主要由黏附分子介导。黏附分子是介导细胞与细胞、细胞与基质间黏附作用的一类分子,大多为糖蛋白。它们参与细胞的生长及分化、细胞的伸展和移动、细胞间信号转导、肿瘤转移、创伤愈合等一系列重要生理和病理过程。根据黏附分子的结构特点,将其分为整合素家族、选择素家族、免疫球蛋白超家族、钙黏素家族及其他未归类的黏附分子。正常人表皮主要表达整合素家族和钙黏素家族黏附分子。
整合素家族是一组细胞表面糖蛋白,是由α和β亚单位经非共价键连接而成的异二聚体糖蛋白,其配基结合区能识别相应配体上的精氨酸一甘氨酸一天冬氨酸序列( Arg-Gig-Asp,RGD)。α、β亚单位结合后可决定整合素的结合活力和特异性,其功能也受表达细胞的影响。整合素广泛分布于各种细胞的膜表面,正常表皮中只在基底层角质形成细胞表达,介导角质形成细胞与基底膜之间的黏附。与黏附有关的基底膜或基质成分包括层粘连蛋白(laminin)、纤维粘连蛋白、纤维蛋白原、胶原、脂多糖、硫酸肝素等。正常皮肤表皮主要表达β1和β4整合素。实验表明β1整合素不仅介导细胞黏附及迁移,而且调控角质形成细胞的分层和分化,在毛囊发生、基底膜及半桥粒的形成和结构的维持中亦起重要作用。α2β1、α3β1主要表达于基底层角质形成细胞的侧面和顶面。前者主要是作为Ⅳ型胶原及层粘连蛋白的受体,后者主要是作为纤维粘连蛋白的受体,也是层粘连蛋白及Ⅰ型胶原的受体。α3β1是皮肤基底膜层粘连蛋白5在表皮角质形成细胞上的主要受体,为皮肤发生过程中形成正常基底膜结构所必须,并能调节张力纤维的形成,在大多数黏附转换细胞和肿瘤细胞中高水平表达。α5β1为纤维粘连蛋白的受体,在正常表皮不表达,但可在一些病理过程或培养中的角质形成细胞表达。研究表明,创伤愈合时及银屑病患者的表皮基底上层也可表达整合素。
钙黏素是一个依赖钙的黏附分子家族,每个分子均由一个包含结合钙序列的细胞外结构区、一个跨膜区和一个与细胞骨架相连的细胞质结构区组成。钙离子是稳定其结构、维持其空间构形及抵御蛋白溶解酶降解的基本条件。角质形成细胞表达E、P-钙黏素。E-钙黏素是构成角质形成细胞黏附连接的主要黏附分子,并在体外介导朗格汉斯细胞与角质形成细胞的连接。P-钙黏素的表达限于基底细胞及相邻的基底上层。
皮肤疾病时真皮的变化
真皮微血管变化的机制
皮肤具重要体温调节作用,与皮肤广泛而发育良好的微循环有关。皮肤有两个水平毛细血管丛:上丛位于表皮下1~1.5 mm,供应真皮乳头层;下丛位于真皮和皮下组织交界处。真皮毛细血管由内皮细胞、细胞间黏合物质和周皮细胞组成。正常皮肤的一般血管分布,约100μm直径的小动脉进入真皮下层,该小动脉有内皮细胞、内弹力膜及数层平滑肌细胞。进入真皮下层后分支1-2次。到真皮中层再分支,此时直径约50 μm,有一层平滑肌细胞,斜行排列呈螺旋状,此时无内弹力膜,仅可见少许弹性纤维。血管继续分支,到真皮上部时直径约15μm,失去肌层变成毛细血管。在乳头下有浅层和深层乳头下丛。乳头下丛上面为终末毛细血管。每一乳头内有一直立的毛细血管袢,无肌细胞,有一层连续均质性的基底膜,膜上无孔隙。真皮中、上层细静脉比细动脉多,大都为水平方向行走。细静脉的直径在真皮中、上层为40~60 μm,在深层为100~400μm,肌细胞逐渐增多,到中静脉时有连续的肌层。
血管新生(angiogenesis):在银屑病、瘢痕疙瘩等疾病中,真皮微血管新生明显。导致血管新生的诱导因素依赖于许多血管生长因子,角质形成细胞可能是血管生成细胞因子(VEGF,IL-8)的主要来源。促进血管新生的生长因子包括血管内皮生长因子(VEGF)、血小板衍生的生长因子(PDGF)、血小板衍生的内皮细胞生长因子(PDECGF)、血管生成素、成纤维细胞生长因子(FGFs)、缺氧诱导因子(HIF)、IL-8、整合素和细胞外基质、血小板反应素等。
VEGF是血管发生的有效介质,长期将VEGF转基因至皮肤导致复杂的炎症状态,具有人银屑病的细胞与分子特点,如增生和真皮血管炎症、表皮增厚(棘层肥厚)、角质形成细胞分化异常、特征性炎细胞浸润等。在K14-VEGF转基因鼠,过多的VEGF可能通过诱导血管炎症应答而导致易感银屑病。另外可能存在其他的机制共同促进皮肤表浅血管丛和角质形成细胞的生长。
新生血管的形成过程如下:
- 角质形成细胞产生并释放血管生长因子,扩散至邻近组织;
- 血管生长因子与邻近血管上的内皮细胞特异性受体结合;
- 内皮细胞活化,产生新的分子,包括酶类;
- 酶溶解血管周围基膜上的小孔;
- 内皮细胞开始分化、增生,通过溶解的小孔迁移到病变组织;
- 细胞黏附分子和整合素(αvβ3,αvβ5)促进新生血管萌芽;
- 基质金属蛋白酶(MMP)产生,融解新生血管萌芽顶端前组织,这些组织在新生血管周围改建;
- 萌芽的内皮细胞参与形成血管管腔;
- 单个血管腔连接形成血管环从而使血液能够流通;
- 最后,新形成的血管管壁被平滑肌细胞和周细胞固定,血流开始。
血管畸形:由于胎儿期发育异常或创伤导致异常结构的形成称为畸形,包括解剖结构的异常或功能改变(如贫血痣)。根据受累血管类型的不同,前者又可分为毛细血管性、静脉性、动脉性、淋巴管性或复合性畸形。毛细血管性者包括先天性大理石纹理样毛细血管扩张症、鲜红斑痣(先天性毛细血管畸形)、疣状血管畸形等,以及由于血管扩张导致的病变如蜘蛛痣、静脉湖、毛细血管动脉瘤等。
真皮成纤维细胞与细胞外基质变化的机制
正常皮肤含有胶原纤维、弹性纤维及网状纤维。胶原纤维较细,交织成网;创伤愈合后正常瘢痕中胶原纤维束界限清楚,结构规整,排列与表面皮肤平行,网状纤维明显增多并与胶原纤维嵌合,弹性纤维减少或消失:增生性瘢痕的胶原纤维呈扁平状,界限不清楚、排列不规则、长短不一、有较多短纤维,有旋涡状或结节样结构,但大多数纤维仍与表面皮肤平行;瘢痕疙瘩中胶原纤维粗大,散乱而不成束状,排列极不规则,与表面皮肤不平行,含有较多粗大、脆性的透明样胶原纤维,有较多结节样结构。
皮肤创伤修复依赖于细胞与细胞外基质(extracellular matrix,ECM)的相互作用。成纤维细胞是主要修复细胞,在某些趋化因子的作用下,由创周向创面移位,并分泌大量的ECM如胶原蛋白(collagen)、纤维连接蛋白(fibronectin,FN)、层连蛋白(laminin,LN)、体外粘连蛋白(vitronectin,VN)、蛋白多糖(proteoglycans,PG.主要有玻璃酸、硫酸软骨素、硫酸皮肤素等)等。成纤维细胞-ECM相互作用时,一方面成纤维细胞增殖,合成分泌ECM填充缺损;另一方面,ECM起着支架和连接作用,并调节成纤维细胞的发育、移位和增殖。在某些细胞因子作用下,成纤维细胞过度增殖,致ECM异常沉积。ECM的降解主要是由肥大细胞、巨噬细胞、内皮细胞及成纤维细胞分泌的胶原酶、糖蛋白酶及其他蛋白酶来完成的。成纤维细胞过度合成胶原、纤维连接蛋白及糖蛋白,以及这些基质成分的降解、塑形不足,均可以导致病理性瘢痕的发生。
整合素介导成纤维细胞-ECM间的相互黏附:成纤维细胞-ECM相互作用是由黏附分子介导的,整合素(integrin)是其中的主要家族。整合素α3β1、α4β1、αvp3和αvβ1能与纤维连接蛋白结合;整合素αvβ1、αvβ3则是VN受体。αvβ5还能调节细胞表面结合的VN的分化与退化,而αvβ3则无此功能。αvβ5的这种功能有利于ECM的塑形,因为二者接触处是相对的紧密部位,成纤维细胞快速向创面迁移需要aαvβ5来调节不同的细胞内信号传导途径。
ECM对成纤维细胞功能的影响:FN及其片段能促进成纤维细胞增殖,使停止生长的成纤维细胞重新合成DNA,进入细胞周期。而对LN及其片段的研究表明,LN短臂内部杆状片段含丰富的半胱氨酸,具有“EGF”样重复单位,该结构在其他几种ECM中也存在,在空间结构上有利于蛋白质相互作用。FN对成纤维细胞、巨噬细胞具有趋化作用,且依赖于FN-成纤维细胞间的黏附,通过此黏附作用,细胞膜蛋白和磷脂进行转甲基反应,细胞骨架中微丝微管收缩,使细胞迁移。Ⅰ、Ⅱ、Ⅲ型胶原及其降解产物、胶原酶降解片段对成纤维细胞均有趋化作用。真皮基质中的蛋白多糖(proteoglycans,PG)可抑制成纤维细胞功能,其中含量较多的是硫酸软骨素B,包括DS-PG Ⅰ(biglycan)和DS-PGⅡ(decorin.核心蛋白多糖,一种小分子量糖蛋白)。增生性瘢痕中的蛋白多糖成分中,decorin仅为正常皮肤中其含量的25%,而硫酸软骨素及双聚多糖为正常皮肤的6倍。decorin在培养细胞中可抑制转化生长因子β(TGF-β),从而推测decorin对成纤维细胞的抑制作用是通过TGF-β完成的。胶原分泌到细胞外,其裂解产物前胶原肽在转录和翻译水平负反馈抑制成纤维细胞合成胶原。创面愈合后,这种负反馈抑制可能减弱乃至消失,使ECM过量沉积而形成增生性瘢痕。在增生性瘢痕和瘢痕疙瘩中,纤维连接蛋白(FN)、蛋白多糖的含量均有明显增加。瘢痕疙瘩中黏蛋白状基质成分则更加丰富,尤其是在其结节状结构中。
病理性瘢痕中细胞外基质代谢的调控:细胞外基质的代谢是一个连续、复杂的过程,受多种因素的调节。其中任何一个或多个环节调节失衡,均可以导致病理性瘢痕的产生和发展。细胞外基质的合成主要受纤维源性细胞因子的调控,包括血小板源性生长因子PDGF、胰岛素样生长因子-Ⅰ(IGF-Ⅰ)、转化生长因子-β(TGF-β)及碱性成纤维细胞生长因子(basic fibroblast growth factor,bFGF),其中最受瞩目的是TGF-β。TGF-β通过增加胶原、纤维连接蛋白、糖氨多糖的合成,增加蛋白酶抑制剂以减少蛋白酶的作用,从而加速组织修复。其主要作用是促使胶原成分的大量合成。但当严重创伤、反复感染等使TGF大量分泌、持续存在时,便会引起细胞外基质的过度沉积,导致病理性瘢痕的发生。PDGF通过刺激巨噬细胞及成纤维细胞的大量增殖,诱导其他细胞因子的释放,扩大急性炎症反应,并直接刺激糖氨多糖的大量生成。
真皮白细胞浸润的机制
白细胞浸润是一个主动的过程,在白细胞和内皮细胞膜上的有关分子相互作用下,经过附壁、黏着、游出和趋化等作用阶段,白细胞才能游出血管到达炎症灶部位。白细胞募集到炎症部位的一系列连续步骤由细胞因子、趋化因子和黏附分子组成的复杂网络介导。
白细胞募集的第一步是从血管腔内自由流动过渡到沿管壁内皮滚动。炎症部位的血管在信号因子作用下,血管扩张。随着血管的扩张,血管的通透性增加,血流变得缓慢。此时,血流中的白细胞离开轴流,开始沿着血管内壁滚动,继而发生白细胞贴在血管内壁的现象,称为附壁,主要由黏附分子家族的选择素介导。活化的内皮细胞表达P选择素(CD62P)和E选择素(CD62E),E选择素的特异性单克隆抗体不能减轻银屑病的表现,而低选择性复合物如efomycine M.通过抑制E选择素和P选择素具有显著的抗银屑病效果。
表达E选择素的内皮细胞与表达在T细胞上的特异性配基相互作用,这些配基为穿膜糖蛋白,含有特殊的碳水化物部分,称为sialyl-Lewisx,定位于皮肤的T细胞表达CLA+sialyl-Lewisx。
滚动的白细胞被趋化因子激活后,通过B2整合素(如LFA-1)或αLβ2整合素与免疫球蛋白超家族黏附分子如ICAM-1(CD54)的相互作用,紧紧地黏附于血管内皮上。另外还有β1整合素与其配基α4β1以及血管细胞黏附分子1的相互作用。
黏着在两个血管内皮细胞连接处的白细胞,接受炎症信号因子后,启动细胞内肌动蛋白的重新组装,以至改变形状,伸出伪足,整个白细胞逐渐以阿米巴运动方式从内皮细胞缝隙逸出,到达内皮细胞和基膜之间,最终穿过基膜到血管外。用电子显微镜可追踪此游出轨迹。—个白细胞通常需2~12分钟才能完全通过血管壁。中性粒细胞、单核细胞、淋巴细胞、嗜酸性和嗜碱性粒细胞都是以此种阿米巴运动方式游出的。血管壁受严重损伤时红细胞也可漏出,但这是个被运过程,是流体静压力把红细胞沿白细胞游出的途径或内皮细胞坏死崩解的裂口推出血管外。
炎症的不同阶段,游出的白细胞也不同。在急性炎症的早期,中性粒细胞首先游出。48小时后组织内则以单核细胞浸润为主,其原因首先在于中性粒细胞的寿命短,经24-48小时中性粒细胞崩解消失,而单核细胞在组织内存活时间长;中性粒细胞停止游出后,单核细胞仍可持续游出;第三个因素为在炎症的不同阶段所激活的趋化因子不同。现已证实中性粒细胞能释放单核细胞趋化因子,因此中性粒细胞游出后必然引起单核细胞的游出。此外,由于致炎因子不同,渗出的白细胞也不同:常见的葡萄球菌和链球菌感染,以中性粒细胞渗出为主;病毒感染以淋巴细胞为主;在一些过敏反应,则以嗜酸性粒细胞渗出为主。游出的白细胞在趋化物的作用下,主动地游向炎症灶部位。
我们对白细胞的渗出过程了解得比较清楚,但对随后通过皮肤细胞外基质的迁移以及决定白细胞在表皮最后的定位和捕获过程仍然知之甚少。
真皮肉芽肿形成的机制
以在炎症局部形成主要由巨噬细胞增生构成的境界清楚的结节状病灶为特征的慢性炎症,称为慢性肉芽肿性炎症。结节较小,直径一般为0.5~2 mm,这是一种特殊类型的慢性炎症。不同的病因可以引起形态不同的肉芽肿。皮肤疾病肉芽肿分为两类:感染性肉芽肿和非感染性肉芽肿。感染性肉芽肿病因比较明确,常见的病原体有细菌、梅毒螺旋体、真菌、寄生虫等,其中分枝杆菌感染多见。非感染性肉芽肿多与感染因素无关,但近来的研究发现,感染因素亦在非感染性肉芽肿的发病中发挥一定的作用,二者没有绝对的界限。这里以皮肤结核肉芽肿和结节病肉芽肿的形成为例阐述肉芽肿形成的机制。
皮肤结核肉芽肿的形成机制:肉芽肿的形成和维持对控制分枝杆菌的感染是必需的,但矛盾的是肉芽肿也导致了分枝杆菌感染所致的典型病理改变。自然界有70多种分枝杆菌,在人类感染的主要是结核分枝杆菌和麻风杆菌。分枝杆菌生长缓慢,逃避了巨噬细胞的吞噬和宿主的细胞免疫应答。感染了分枝杆菌的树枝状细胞激活CD4+和CD8+T细胞,被募集至感染部位激活感染的巨噬细胞。同时,结核杆菌阻止了吞噬溶酶体对感染吞噬小体的融合和酸化,通过IFN-γ部分抑制巨噬细胞的活化,最终导致一些结核杆菌在感染部位持续存在而形成一慢性刺激性抗原,T细胞在巨噬细胞周围逐渐积聚。在细胞因子的刺激下,巨噬细胞分化为上皮样细胞,并相互融合形成典型的巨细胞而逐渐形成肉芽肿。肉芽肿形成后,分枝杆菌的死亡和存活处于一平衡状态。这种限局性结构导致淋巴细胞和巨噬细胞处于更紧密的位置以有利于灭活分枝杆菌。但是,某些逃避巨噬细胞杀伤作用的结核杆菌潜伏下来而被肉芽肿包裹。
表达αβT细胞受体(TcR)的CD4+T细胞的活化在分枝杆菌肉芽肿的形成过程中发挥重要作用。其他细胞如表达αβTcR的CD8+T细胞、表达γδTcR的T细胞以及CD1限制性CD4/CD8双阴性T细胞均发挥一定的作用。CD4+T细胞在结核杆菌感染的树突状细胞的刺激下分化形成Thl样CD4+T细胞,分泌IL-2、IFN-γ和淋巴毒素lymphotoxin-α(LTα),上调MHCⅡ类分子、协同刺激分子CD80和CD86.促进IL-12和炎症性细胞因子的分泌。
5个基因与分枝杆菌感染有关:IFN-γR1、IFN--γR2、Stat1、IL-12 p40和IL-12 Rβ1。IFN--γ信号途径缺陷导致个体对结核杆菌易感,而且不能形成肉芽肿,说明IFN-γ对激活巨噬细胞和肉芽肿形成是必需的。另外,IL-12和IL-23在产生Th1样细胞免疫的过程中非常关键。
结节病肉芽肿的形成机制:结节病肉芽肿的形成需要抗原提呈细胞与T细胞之间的相互作用。这种免疫应答发生于一具有遗传易感性的患者,疾病的严重程度取决于其易感基因(如HLA和TNF)。在肉芽肿形成的初期,巨噬细胞接触激发抗原,但却不能吞噬(吞噬障碍)。抗原经经典的MHCⅡ限制途径传递给抗原特异性CD4+T细胞。活化的巨噬细胞募集单个核细胞(主要是单核细胞和CD4+T细胞)。这些细胞在炎症部位聚集,试图阻止抗原或病原体的入侵。接着,在趋化因子的作用下,炎细胞聚集到肉芽肿部位。在效应阶段,肉芽肿形成,特异性细胞被募集到炎症部位,主要是CD4+T细胞。但是,如果最初的抗原刺激不能形成肉芽肿,则嗜酸性粒细胞和中性粒细胞浸润至炎症部位。小腿肉芽肿性炎症的持续或纤维化取决于炎细胞、调节细胞、凋亡和Th1/Th2细胞因子之间的精细平衡。调节性细胞因子如TGF-β和IL-10与炎症性细胞因子TNF-α和IFN-γ在局部达到平衡,但若TGF-β相对过多则导致纤维化。
表皮-真皮交界处的变化
表皮与真皮交界处的结构即皮肤基底膜带(basement membrane zone,BMZ),PAS染色阳性。BMZ很窄,仅50~100 nm厚,可分为4层:
- 半桥粒:为与真皮相邻的基底细胞质膜,其上有对表皮真皮连接起关键作用的半桥粒;
- 透明板:位于基底细胞质膜的下方;
- 致密板;
- 致密板下带。
在BMZ的表皮侧,可见一些电子致密区—半桥粒,它将表皮连接到真皮上起着重要的作用。
BMZ的结构
半桥粒(hemidesmosome):位于表皮基底细胞层的下方,含有桥粒的一半结构:单个的胞质内黏附板,胞膜内侧的半桥粒斑与胞质内张力丝相连接,其上有BP230(大疱性类天疱疮抗原BPAg1)、BP180(BPAg2)、亲和素和网蛋白等。半桥粒结构的维持受局部环境Ca2+度影响。BP230是胞质内蛋白,其C端在胞质内,深入胞内约75 nm,与细胞内的张力丝相互作用,因此和细胞内的骨架系统连成一体。BPAg1相对分子质量为230 kD,属于斑蛋白家族。它与表皮基底细胞内另一个结构蛋白网蛋白都位于半桥粒的内板,是半桥粒内板的主要成分。剔除BPAg1基因的转基因鼠显示,BPAg1在与基底细胞中由角蛋白所组成的张力细丝的连接上起着重要的作用。该鼠半桥粒的内板缺如,使与半桥粒相互连接之张力微丝发生彼此分离,张力微丝退缩至核周围。但这个突变结果并不导致表皮真皮的分离,也不形成表皮内水疱。参与这一作用的还有亲合素P4亚单位,其在胞膜外侧有50个氨基酸残基,在胞质内侧有1 000个氨基酸残基,人们推测其在细胞内骨架系统及细胞与基底膜的信息交流中发挥重要作用。亲和素是黏附分子家族的重要成员,在细胞间的黏着、迁移和分化中起着重要作用。BPAg2是在大疱性类天疱疮和妊娠疱疹发病中起主要作用的抗原。BPAg2是一个跨膜蛋白,相对分子质量为180 kD。它小部分位于基底细胞半桥粒的外板,大部分则跨越基底细胞膜的真皮侧,向下延伸,穿过透明板一直到达致密板,可能参与锚纤维的组成。在基底细胞内的是BPAg2的氨基端,细胞外部为BPAg2的羧基端。BPAg2的胞膜外区还包含一个由16个非胶原区(NC16A)分隔开的胶原区,NC16A位于临近穿膜区的部分。BPAg2的细胞外部分与半桥粒的另—个跨膜蛋白——整合素α4β6协同,共同使基底细胞黏附在基底膜上。与BPAg1比较,BPAg1位于基底细胞内,而BPAg2位于细胞外,它将首先与血清中的自身抗体发生特异结合,因此认为在大疱性类天疱疮(BP)的发病中,BPAg2起着更为重要的作用。
编码BPAg2的基因COL17A1基因突变与一个新发现的大疱性表皮松解症亚型——泛发性萎缩性良性大疱性表皮松解症有关。另外,BPAg2不但是BP、妊娠疱疹,而且也是线状IgA大疱性皮肤病的自身抗原。
另外,整合素家族和网蛋白也位于此区。整合素已在第五章第三节介绍过。网蛋白(plectin)属于斑蛋白家族,是相对分子质量为518 kD的大分子,是多功能细胞骨架相关性蛋白,在多种组织中有表达,包括上皮和间质中都有表达。它在基底细胞中形成二聚体,与BPAg1一起位于半桥粒的内板,与角蛋白中间丝相连接,起着将基底细胞通过半桥粒连接于基底膜,并有稳定半桥粒内板的作用。网蛋白具有异质性,在基底细胞半桥粒中的网蛋白为DH1。编码网蛋白的基因PLEC1定位于8q24。该基因突变可导致伴迟发性肌肉萎缩的大疱性表皮松解症。
透明板:与半桥粒及致密板相比,透明板的电子密度较低,其主要成分是板层素及其异构体,它们组成了细胞外基质和锚丝。锚丝从角质形成细胞的基底面通过透明板达到致密板。在锚丝中,板层素是其主要组成成分。板层素受体作为亲合素家族的配体,广泛地存在于细胞外基质中,由3个亚单位组成,位于BMZ上的板层素主要是板层素1、5和6。
致密板:电子显微镜下此层厚35~45 nm。有学者认为致密板为真正的基底膜。构成此层的物质主要是Ⅳ型胶原、基底膜蛋白多糖(硫酸乙酰蛋白多糖),也有板层素6的存在。构成透明板的板层素5,不仅位于透明板下部,也可见于致密板。Ⅳ型胶原分子是由两条相同的α1链和一条α2链组成的柔软线状蛋白质分子,长约400 nm。Ⅳ型胶原分子通过自体间的相互交联,形成连续的三维网格是稳定基底膜的重要支持结构。
致密下层:在致密下层中有锚纤维通过,把致密板和其下方的真皮连接在一起。Ⅶ型胶原是构成锚纤维的主要成分,属于胶原家族,由3条前α链组成的三螺旋结构,分子量约290 kD,由角质形成细胞和成纤维细胞合成和分泌,它与Ⅳ型胶原或锚斑结合,并与真皮纤维交织在一起,维持表皮细胞与结缔组织的固着。Ⅶ型胶原不同于Ⅰ-Ⅳ型胶原之处在于在胶原的三螺旋结构中有一些非胶原区的插入,这增加了它的可伸屈性。Ⅶ型胶原通过C端重叠形成反向二聚体,由二硫键彼此连接。大量的二聚体组成锚纤维。锚纤维的N端与基底膜致密板的锚斑和层粘连蛋白5相连接。编码Ⅶ型胶原的基因COL7A1位于3p21.1,由118个外显子组成,基因全长32kb,是目前发现的最大基因之一。Ⅶ型胶原是获得性大疱性表皮松解症的自身抗原。COL7A1基因突变与营养不良性大疱性表皮松解症的发病有关。
与BMZ相关皮肤病的发病机制
人皮肤BMZ是自身免疫性疾病的靶点。
针对半桥粒绣明板上层的自身抗体导致的皮肤病:
- 大疱性类天疱疮(BP):大疱性类天疱疮是最常见的自身免疫性表皮下大疱性疾病,主要见于老年患者。BP患者皮损周围外观正常的皮肤进行直接免疫荧光(DIF)检测,在80%患者见IgG和C3的沉积。BP循环抗体的靶抗原是位于半桥粒上的两个重要的生物大分子:BP230(BPAg1)和BP180( BPAg2)。免疫印迹研究证实大部分BP血清识别的靶抗原是BP230,部分BP血清识别或同时识别BP180抗原。目前认为BP180抗原在BP的发病中起着始动作用。
- 线状IgA大疱性皮病(linear IgA bullous dermatosis,LAD):LAD是以DIF见IgA在皮损周围皮肤的BMZ线状沉积而命名的表皮下大疱性皮肤病。有关LAD的靶抗原的研究较多,免疫印迹研究发现LAD自身抗原存在着明显的异质性,不同的LAD患者体内循环IgA型BMZ抗体结合的靶抗原分子量不相同。其靶抗原可以是BMZ中的BP230、BP180.相对分子量为97 kD、285 kD、255 kD的抗原和Ⅶ型胶原等。免疫电镜对其定位研究发现,抗原可定位于半桥粒上、透明板中、致密板和(或)致密下层等。
针对透明板下层自身抗体导致的疾病:
- BPAg自身抗体性瘢痕性类天疱疮(cicatricial pemphigoid,CP):CP系免疫调节缺陷而导致自身抗体的产生,沉着于基底膜引起炎细胞趋化、脱颗粒、成纤维细胞活化、瘢痕化与纤维组织炎。一般认为CP的靶抗原为相对分子质量45 kD的表皮提取物多肽,但也有研究认为部分CP的靶抗原为BP180、相对分子质量为105 kD的基板蛋白。CP抗体识别BP180氨基端多肽区域,免疫电镜证实CP患者血清识别的抗原表位位于透明板下部和致密板上。
- 抗板层素瘢痕性类天疱疮:最主要的自身抗原之一基底膜糖蛋白的层粘连蛋白。板层素5和板层素6可被一组CP循环抗体IgG所识别,进一步研究发现此组CP患者血清抗体IgG主要识别相对分子质量为60 kD的抗原(板层素6)和相对分子质量为40 kD的抗原(板层素5)。因此命名为抗板层素CP。免疫电镜见该抗体的结合部位在透明板下部。基底膜损害可能是蛋白酶介导而引起的细胞脱颗粒,使真皮与表皮分离。亦有认为本病为低白介素-6及高滴度的肿瘤坏死因子所致。
- 抗β105类天疱疮:DIF检测见患者皮损中有IgG在BMZ线状结合,免疫印迹证实该自身抗体的靶抗原为相对分子质量105 kD的抗原。金标免疫电镜研究显示它位于BMZ的透明板下部。对这种靶抗原进一步研究揭示其在N端和肿瘤相关抗原有同源序列,但与BMZ的其他成分如板层素等没有同源性,推测这种物质可能由角质形成细胞和成纤维细胞所产生。
针对致密板下层自身抗体导致的疾病:
获得性大疱性表皮松解症(EBA)是由IgG所介导的一种表皮下水疱形成的皮肤黏膜疾病。DIF在皮损周围外观正常的皮肤组织中见IgG在BMZ处呈一宽的线状结合带,盐裂皮肤的DIF见IgG结合在真皮侧。免疫印迹和免疫沉淀揭示循环抗体IgG识别相对分子量290 kD的真皮提取物多肽,即Ⅶ型胶原。现已发现EBA的自身抗体所识别的表位在Ⅶ型胶原的N端的非胶原样区(NC1)。
疾病时皮下脂肪组织的变化
皮下脂肪组织是皮肤最深、最厚的一层,主要由脂肪细胞组成,也含有纤维结缔组织——胶原和弹性硬蛋白。成群的脂肪细胞组成小叶,每个小叶由结缔组织包裹,后者形成小叶间隔。结缔组织纤维之间有基质,含有血管、淋巴管、神经和各种细胞。
仅1/3的脂肪组织含有成熟的脂肪细胞,另外2/3由小血管、神经、成纤维细胞以及脂肪前体细胞组成。成熟的脂肪细胞包括两种类型:白色脂肪细胞和棕色脂肪细胞。这两种细胞的颜色和功能不同,白色脂肪组织(WAT)为黄色或象牙白色,主要含有白色脂肪细胞。棕色脂肪组织(BAT)主要包含棕色脂肪细胞。
白色脂肪细胞含有一大的“单房性”脂滴,体积超过50 μm。白色脂肪细胞呈椭圆形,这样可以在最小的空间内达到最大的体积,其直径大小不二,约30-70 μm。脂滴占据了细胞内的绝大部分空间,将细胞质和细胞核挤压在一边。棕色脂肪细胞形成多个小的“多房性”脂滴,其特征是含有大量线粒体,细胞为多边形,细胞核位于中央。棕色脂肪细胞体积相对白色脂肪细胞小,约20-40μm。
所有的棕色和白色脂肪组织均接受血管供应和神经支配。与WAT相比,BAT血管更丰富,毛细血管更致密。这种广泛的血管化以及致密的线粒体结构形成了BAT的棕色。WAT和BAT均受去甲肾上腺素能神经纤维支配,这种神经纤维在WAT主要局限于毛细血管壁,而BAT则通过突触直接作用于棕色脂肪细胞的细胞膜。
皮下脂肪组织分为浅部皮下脂肪组织和深部皮下脂肪组织,主要是WAT,其功能主要与脂质代谢、糖代谢和内分泌有关。BAT则将能量转换为热量,参与体温调节。
近年来的研究发现,脂肪细胞除以上功能外,也具有内在的炎性本质。像巨噬细胞一样,脂肪细胞对微生物的感染以及细胞因子介导的炎症信号非常敏感,它们表达一多种受体以感知病原体和炎症。受体受到刺激后激活多种炎症信号转导级联反应,脂肪细胞分泌多种炎症因子激发急性炎症反应。脂肪细胞对肿瘤坏死因子α(TNF-α)非常敏感,后者通过其p55和p75 TNF受体活化NF-κB、细胞外信号调节激酶、p38丝裂原活化的蛋白激酶、PI-3激酶以及Jun-N端激酶级联体。脂肪细胞还表达TLR4,当受到内毒素的攻击时,通过TLR4激活p65/p50和p68/p52 NF-κB信号传导途径。反过来,这些途径又可诱导炎症介质如IL-6、血清淀粉蛋白A3和TNF-α的表达。
在感染和炎症信号的作用下,脂肪细胞诱导炎症急性阶段介质的表达和分泌,包括TNF-α、纤溶酶原活化抑制因子-1、11-1β、IL-6、IL-8、IL-10和IL-15、肝细胞生长因子、补体等。这些脂肪细胞分泌的细胞因子对促进炎症的发展发挥重要的作用。
