
大疱性表皮松解症(Epidermolysis Bullosa、EB,以下统一简称EB)是指皮肤或黏膜受到轻微外伤即可引起水疱的一组异质或多相的遗传性皮肤病,有时也称为“机械性大疱性皮肤病”。这是一组较罕见的疾病,美国在1990年统计,所有类型EB的平均发病率为百万人口8.22人。EB的发生与地域和种族之间关系不大。
本组疾病包括三个主要类型,即单纯型EB、交界型EB和营养不良型EB,共可分为至少20多种临床类型。角蛋白5和7、层连蛋白232、Ⅶ型胶原、大疱性类天疱疮抗原2、网格蛋白、α6β4整合素等7个靶蛋白中任何一个基因突变均可导致一种EB的发生。虽然这类疾病非常少见,但对其病理生理的研究,使我们对角蛋白、胶原和细胞处基质的细胞及分子生物学作用有了更多的了解。同时涉及的上皮细胞的黏附、迁移和分化,基底膜带的生物学作用的基础研究也促进了皮肤科基因治疗的发展。
目前公认的常见及少见的EB类型及亚型见下表:

病因及发病机制
所有类型的EB,其特征性的表现为皮肤对机械力脆性增加。原因涉及10多个编码结构蛋白中的基因突变,这些蛋白通常位于真皮、真表皮交界处或者真皮乳头上层。发生突变的蛋白在皮肤的分布决定EB大疱的位置。
- 单纯型EB:绝大多数单纯型EB是常染色体的显性遗传。总体来说,单纯型EB的相关靶蛋白是角蛋白5(K5)和角蛋白14(K14)基因突变。这些角蛋白位于表皮的基底层。已经确定与之有关的EB亚型包括:Weber-Cockayne,Koeber,Dowling-Meara以及伴斑驳色素的单纯型EB。泛发性EB伴肌营养不良患者例外,是基底细胞半桥粒中的一种网格蛋白(plectin)基因发生突变。
- 交界型EB:所有类型的交界型EB(JEB)是常染色体隐性遗传疾病。JEB多见于3个蛋白之一的失功能(继发于基因突变)。最重的类型JEB-Herlitz是由于编码laminin332的3个蛋白亚单位基因的复合杂合子突变,该蛋白是真表皮连接处透明板的主要蛋白成分。轻型JEB(JEB-non-Herlitz),以前称为泛发性萎缩性良性JEB,是由于laminin332或大疱类天疱疮抗原-2(bullous pemphigoid antigen-2)(已知为X Ⅶ型胶原蛋白)。合并幽门闭锁的JEB是由于编码整合素α6β4的两个基因之一发生突变。
- 营养不良型EB:营养不良型EB既可以是常染色体显性又可以是常染色体隐性遗传方式。显性营养不良型EB(DDEB)是由于Ⅶ型胶原蛋白发生负显性突变。通常这一突变发生在蛋白中糖基结合位点,并不导致Ⅶ型胶原分子的截断。虽然突变的蛋白可导致皮肤的结构异常(皮肤的脆性增加,锚纤维数量和发育异常,疱发生在致密板下等),但是免疫组织化学染色显示DDEB的真表皮与正常皮肤无明显差异。
隐性营养不良型EB(RDEB)通常是由于Ⅶ型胶原蛋白的复合杂合子突变。提前出现的终止密码子可导致蛋白的截断,Ⅶ型胶原不稳合成。在最重型隐性EB,如Hallopeau-Siemens RDEB。由于Ⅶ型胶原发生突变,通常在皮肤活检标本中难以检测到锚纤维,同时抗Ⅶ型胶原蛋白分子主要表位的抗体也不能在免疫组织化学中检测到该蛋白。
有些RDEB患者确实保留有Ⅶ型胶原蛋白的特定的片段(非胶原域的氨基酸末端NC1),这一突变可能增加皮肤鳞状细胞癌的易感性。在某些少见类型的显性营养不良型EB,如见于新生儿的暂时性大疱性表皮松解,发生于Ⅶ型胶原蛋白基因突变,但是疱通常仅发生于出生后的1~2年。当临床大疱形成时,正常的Ⅶ型胶原蛋白在真表皮交界处线状沉积,这说明这些蛋白从角质细胞胞质中分泌出来到细胞外基质中,存在运输和沉积的短暂延迟。
EB各类型及其相关靶蛋白、超微结构,以及皮肤发生裂隙、水疱位置比较见下表。

临床症状
遗传性EB特征性表现为皮肤脆性增加,皮肤受到轻微的摩擦即可导致皮肤的分层,通常在数分钟内发生紧张的水疱、糜烂和结痂。
瘢痕(几乎都是萎缩性)可发生于任何类型和亚型的EB,包括单纯型EB。局限性单纯型EB患者的瘢痕发生率大约仅15%,然而RDEB患者几乎均发生瘢痕。别的皮肤症状在所有的主要类型和亚型EB中的发生率变化不明显,如甲萎缩和缺失、粟粒疹和头部瘢痕性脱发。
网状色素沉着斑可见于少见的单纯型EB,常称为斑驳色素型单纯EB(EBS-MP)。表皮剥脱见于单纯性表浅性EB(EBSS),本亚型缺乏明显的水疱。呈弧形或多环形排列的群集水疱通常见于泛发性EBS亚型,称为“疱疹样EBS”(Dowling-Meara,EBS-DM),EBS-DM可特征性进行性出现掌跖角化病。JEB的Herlitz亚型的特征表现有过度增生的肉芽组织,累及腔口周围、关节伸侧、上背部和颈项部位皮肤。
JEB或RDEB反式亚型患者的疾病初期表现为皮肤褶皱部位擦烂。在某些少见的局限性JEB类型患者,皮损最常见于肢端部位。向心性RDEB,特征性表现为肢端部位的水疱缓慢向躯干部位发展。
主要类型的EB亚型可以累及患者的皮肤,也可以特征性累及其他的上皮,包括眼结膜、口腔、消化道(除了胃)和生殖道。这些特异部位的受累多见于营养不良型EB,较少见于JEB。表现为大疱、糜烂、溃疡和(或)瘢痕。当眼部受累后,可导致新生血管形成和失明。累及食管,如果为慢性和持久性,可形成瘢痕、缩窄,甚至更少见的完全闭锁。小肠受累可导致慢性吸收不良。而累及大肠远较小肠少见,前者可伴有便秘和狭窄。重度受累的患者可发生食管反流。泌尿道复发性水疱可导致尿道缩窄,后者如果持续存在可能导致尿反流和肾积水。有些JEB伴幽门闭锁的患者可发生消化道和肾脏病变。
牙釉质发育不良是所有JEB类型的共同特征。临床上表现为乳牙和恒牙表面的缺失。如果未予处理,可导致儿童时期就发生牙齿缺失。
原发性假性蹼指(手和脚趾畸形)可见于RDEB,尤其是重度泛发性亚型(Hallopeau-Siemens,RDEB-HS),虽然也见于DDEB和JEB患者。最初表现为指近端网孔样融合,如果未经治疗或者病情进展,指端形成融合,被瘢痕组织包围。双手活动性差,而后出现骨质吸收,肌肉萎缩,最终双手完全致残。
骨质疏松是重症泛发性EB的一种共同表现,特别是在RDEB-HS和交界型EB更明显。
泛发性单纯型EB亚型,由于网格蛋白缺乏,出现轻度到重度的肌营养不良。肌肉症状可初发于婴儿期或稍晚,先天性幽门闭锁是发生于泛发性交界型EB的一种罕见亚型(JEB-PA)。
少数严重的泛发性EB患者会出现慢性肾衰竭竭,特别是在RDEB-HS患者,可以致死。少数严重的泛发性EB,特别是RDEB患者,可以发生心肌病。硒元素缺乏可能是发病因素,尽管目前还未得到证实。
生长迟滞在交界型EB婴儿患者会较常见,可致死亡。在出生后6年内的另一个主要死亡原因是由于支气管大疱致急性气管阻塞。
多发性皮肤鳞癌是一个主要的合并症。通常发生在RDEB。肿瘤经常出现于长期未愈合伤口和角化过度的皮损周围。组织学上,分化良好。边界不清楚,很难完全切除,易复发,局部转移和最终远处转移。对化疗和放疗不敏感。多发性皮肤鳞癌是中年和中年后EB患者的主要死因,初诊鳞癌后存活期在5年内。多发生在RDEB(特别是RDEB-HS)和少数交界型EB患者。在RDEB-HS患者,20岁时至少发生一处以上鳞癌的几率是6%,到40岁时高达80%。而在非RDEB-HS患者,同期肿瘤的发生率明显低于RDEB-HS患者。
少数RDEB的儿童患者可以出现恶性黑素瘤,在12岁时,发生率为1.5%,尽管恶性黑素瘤在RDEB是少见的合并症,在儿童期早期需要密切监护。EB儿童患者,特别是那些交界型EB-nH,可以出现较大的外形不规则、色素加深的黑素细胞痣,临床类似恶性黑素瘤,而在组织学上是良性。
各型大疱性表皮松解症的临床表现
单纯型大疱性表皮松解症
单纯型大疱性表皮松解症(epidermolysis bullosa simplx,EBS):
- 泛发型单纯型大疱性表皮松解症:亦称Koebner型(EBS-K),这是EBS中最常见的一型,为显性遗传,有完全外显率,发生率为1/500000。临床特征为手部关节、肘部、膝部、足部和其他易反复损伤的部位发生水疱、大疱和粟丘疹。尼氏征阴性。黏膜和甲不被累及(下图1)。儿童通常在出生时或出生后不久患病,几个月内有所改善,但在开始爬行时和儿童期可能复发,部分患者的水疱可较长期存在和泛发。夏季严重,冬季有所缓解。损害稀少,不会引起严重萎缩。随病程的发展,可消退。患者一般发育和健康状况正常。
- 局限型单纯型大疱性表皮松解症(localized epidermolysis bullosa simplex):亦称Weber-Cockayne型(EBS-WC),该病见于手足部位反复发作性大疱,是常染色体显性遗传,临床原发损害主要是水疱,局限于手足,热天或走路多时可使病情加重,常于7岁后稍见好,可伴掌跖角化。大疱位于表皮内和基底层上,愈后不留瘢痕(下图2)。
- Ogna型单纯型大疱性表皮松解症(EB simplex,Ogna) 为常染色体显性遗传,该亚型仅发生于挪威和德国,可见泛发性青紫和出血性水疱。出血性大疱和拇趾甲弯曲是两个特征性表现。
- 疱疹样型单纯型大疱性表皮松解症(EB herpe-tiformis) 亦称Dowling-Meara型(EBS-DM)。这是一种EBS的常染色体显性遗传变异型。临床表现类似疱疹样皮炎。出生时即可有全身严重水疱,血性,排列成环,愈后不留瘢痕,但可见粟丘疹,如能度过婴儿期至7岁,则可逐渐见好,亦可见掌跖角化过度。可累及口腔黏膜。甲脱落后可再生,有时可见甲营养不良。
- 斑驳色素型单纯型大疱性表皮松解症(EB simplex with mottled pigmentation,EBS-MP) 该亚型表现为儿童早期出现肢端大疱,呈季节性发作。大疱通常为血性。斑纹状色素沉着主要分布在腋下、肢端或下腹,为2~5mm大小的色素斑。以后可以出现点状掌跖角化和甲营养不良。
- 单纯型大疱性表皮松解症伴肌营养不良(EB simplex with muscular dystrophy,EBS-MD) 此型与迟发性神经肌肉疾病伴发,为常染色体隐性遗传。出生时即见泛发性水疱,伴有瘢痕、粟丘疹、萎缩、甲营养不良、牙齿异常、喉蹼和尿道狭窄。进行性肌营养不良伴肌无力甚至肌力丧失发生于儿童期或之后。
- 浅表性单纯型大疱性表皮松解症(epidermolysis bullosa simplex superficialis,EBSS) 为常染色体显性遗传,特点为表皮裂隙发生在角层下,体表糜烂,很像落叶性天疱疮样,愈后留有局部萎缩性瘢痕或炎症性色素沉着。仅一家族报道其发病为Ⅶ型胶原基因突变,提示此病例可能为非典型营养不良型EB,其分子机制尚需进一步研究。
- 致死性常染色体隐性单纯型大疱性表皮松解症(lethal autosomal recessive epidermolysis bullosa simplex,EBS-AR) 1985年Salih报道一家族13名成员出生后不久发生水疱、泛发,不伴有瘢痕或粟丘疹,好发于肢体远端。口腔黏膜受累轻。甲、齿及发不被侵犯。患儿多2岁左右死亡,仅1例存活至成年。电子显微镜观察基底细胞张力微丝很少。另一报道为K14基因突变。
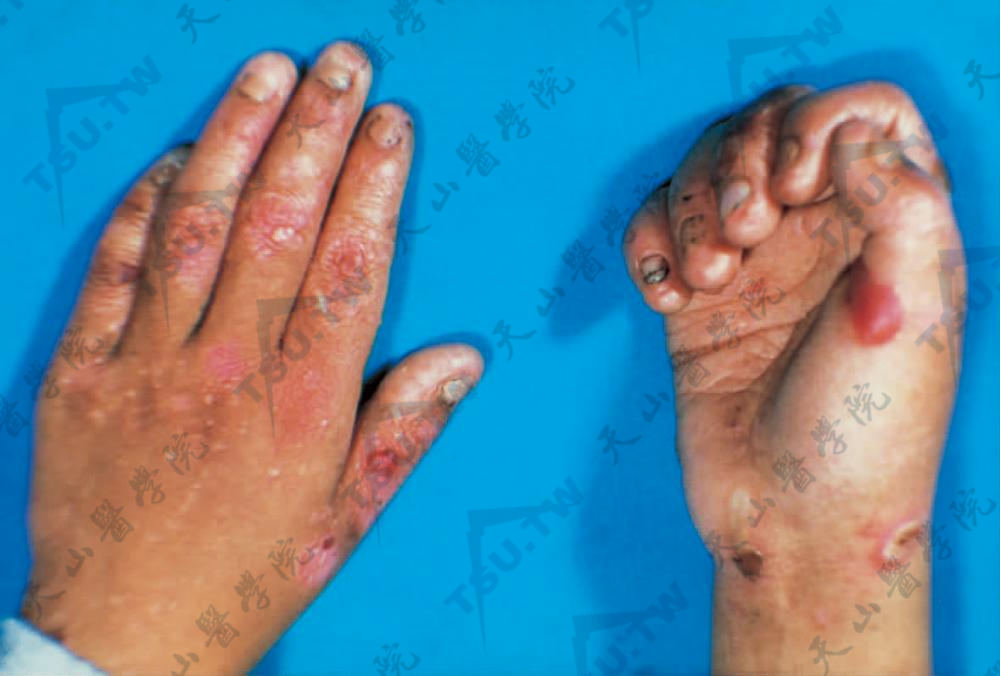
图1:两手部皮肤受摩擦部位见水疱和大疱
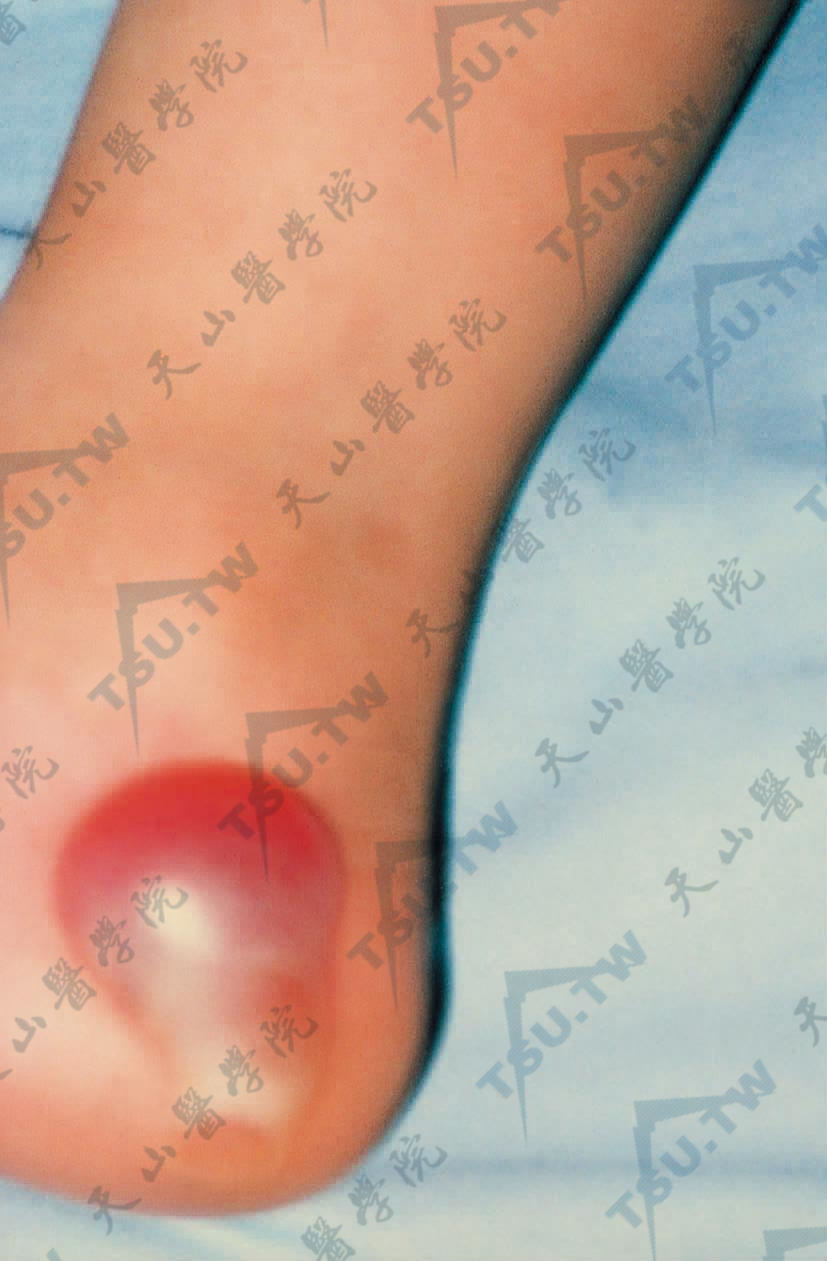
图2:足部因摩擦产生一大疱
交界型大疱性表皮松解症
交界型大疱性表皮松解症(junctional epidermolysis bullosa,JEB)
- Herlitz型交界型大疱性表皮松解症(JEB-H) 又称Letalis型大疱性表皮松解症,为常染色体隐性遗传,大多数出生时可有泛发性水疱,广泛性剥脱,数月内可死亡。手部损害相对少,伴特征性口周和鼻周有肥厚性肉芽组织。糜烂可持续多年。可见甲营养不良或无甲,有时有明显的瘢痕性脱发。常见严重的口腔受累,包括瘢痕和口裂变小。牙齿变形常见。喉部和支气管损害可导致呼吸窘迫,甚至死亡。其他系统性并发症包括肌肉骨骼变形、胃肠道损害、泌尿生殖器和眼部病变。食管累及可以造成食管狭窄。幼儿期发病且存活的患儿,出现生长迟缓,且常见中度至严重的顽固性贫血,多数患儿于2岁内死亡。
- 良性泛发性萎缩性大疱性表皮松解症(generalized atrophic benign epidermolysis bullosa) 即非Herlitz型交界型大疱性表皮松解症(JEB-non-Herlitz,JEB-nH)大多数出生时即出现症状,见泛发性水疱和萎缩、黏膜受累、甲增厚、甲营养不良或缺失。乳牙和恒牙的釉质缺陷和萎缩性秃发是明显的特征。多发性皮肤鳞状细胞癌已有报道。本病为常染色体隐性遗传。与Herlitz型大疱性表皮松解症相反,患者常可存活至成年(下图3、4)。
- 交界型大疱性表皮松解症伴幽门闭锁(junctional epidermolysis bullosa with pyloric atresia,JEB-PA) 这种罕见的常染色体隐性遗传形式的交界型EB,在出生时即出现严重的皮肤黏膜脆弱和胃幽门梗阻。尽管幽门闭锁可修复,但新生儿仍可因严重的皮肤疾病而死亡。如果新生儿期存活下来,水疱可消失。然而,可出现尿道永久性瘢痕,并伴输尿管-膀胱结合部狭窄。
- 瘢痕性交界型大疱性表皮松解症(cicatrical junctional epidermolysis bullosa) 1985年Haber等提出的另一交界型EB。因为水疱愈合形成瘢痕,可导致并指和挛缩,口腔黏膜病变可发生,并伴有前鼻孔狭窄。
- 进行性交界型大疱性表皮松解症 又称神经营养型大疱性表皮松解症(EB dystrophica-neurotrophica),由Gedde-Dahl报道。发病迟,多发生于儿童时期或成人期,常有甲营养不良,水泡发生于手足,以后发展至膝肘。进行性萎缩较早,指纹消失及轻度指痉挛,牙釉质缺陷,舌乳头消失。口腔黏膜有时受侵,还可伴有先天性进行性感知耳聋。超微结构显示透明板增宽,为均质性物质沉积。半桥粒正常,发病机制与分子学基础尚不明。
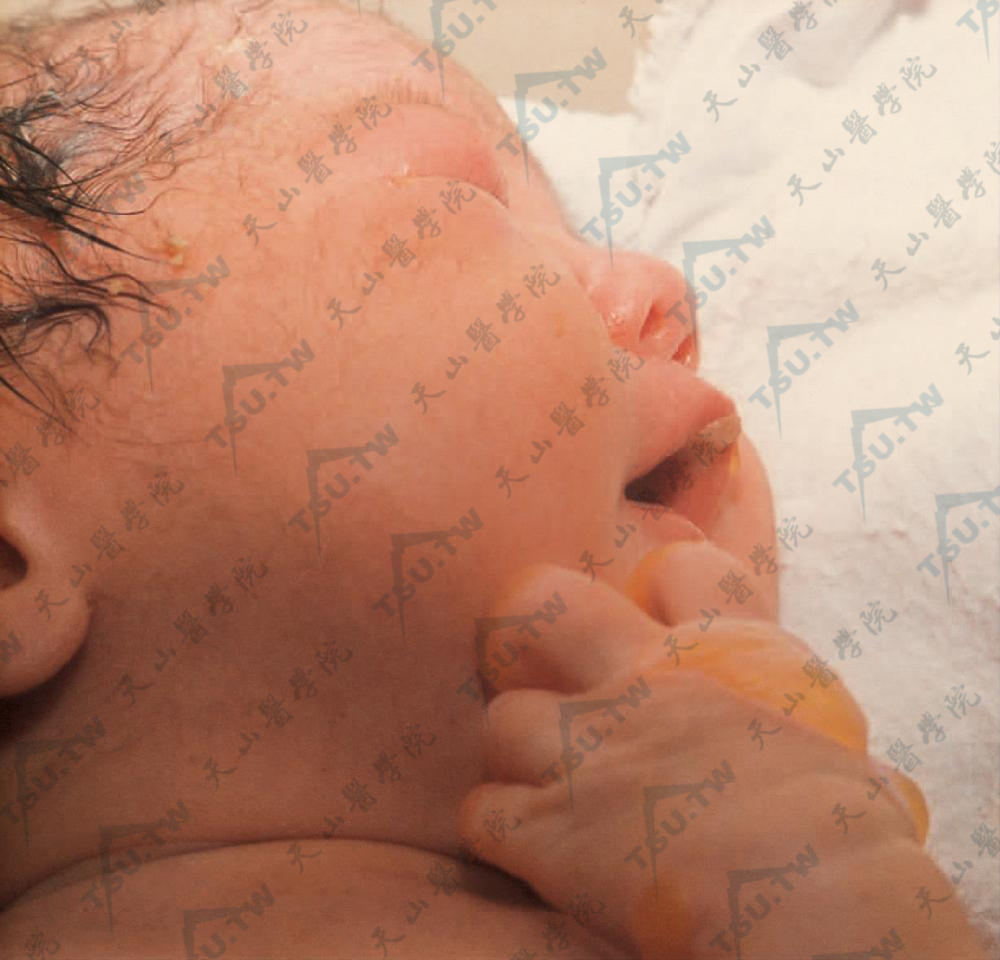
图3:婴儿出生时口周、唇部大疱
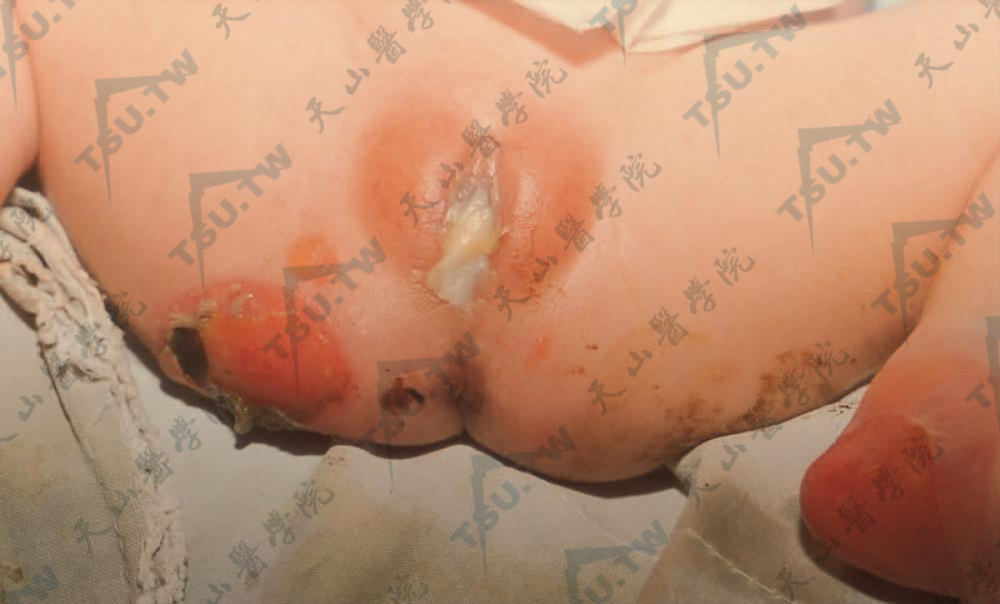
图4:新生儿外阴生殖器部大疱
营养不良型大疱性表皮松解症
营养不良型大疱性表皮松解症(dystrophic epidermolysis bullosa,DEB)
一、显性营养不良型大疱性表皮松解症(dominant dystrophic epidermolysis bullosa,DDEB)
患者通常身材正常,健康状况良好。婴儿早期或儿童期发病,水疱及大疱多位于四肢伸侧,尤其是关节部位,特别是趾、指、踝、肘等关节面上多见。甲可增厚。尼氏征常阳性。皮损痊愈后留有瘢痕及萎缩,有时能形成肥厚型瘢痕和瘢痕疙瘩。在耳轮、手背、臂及腿伸侧常有粟丘疹。常波及黏膜,口腔黏膜、舌、腭、食管及咽喉部可有糜烂。当喉部被波及时,常引起持久性声音嘶哑;在齿龈口唇沟间可有瘢痕挛缩;因咽喉部结瘢痕可致吞咽困难。结合膜常不波及。毛发和牙齿正常(下图)。
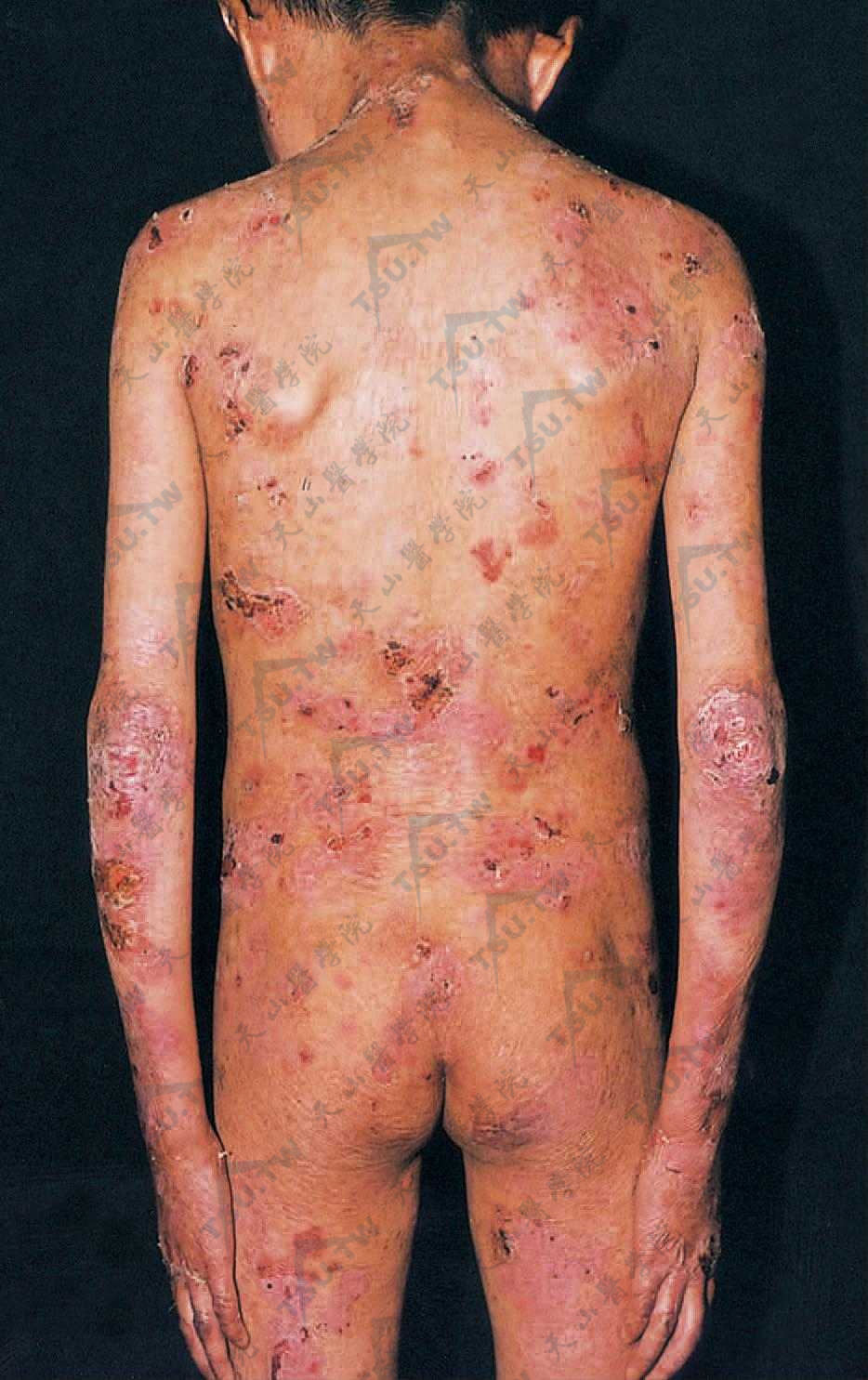
四肢伸侧、颈、腰易受摩擦部位发生大疱及水疱,并可见血疱,愈后留有瘢痕及萎缩
其他改变有甲营养障碍、秃发、体毛缺失、侏儒、爪形手、指骨萎缩及假性并指等。有两个变异型,即白色丘疹样型(Pasini)及增生性变异型(Cockayne-Touraine)。前者是白色丘疹样损害,约数毫米至数厘米大小,可融合成不规则的地图状。多发生在躯干,少数可见于四肢,皮损类似小瘢痕、结缔组织痣或扁平苔藓(下图)。后者皮损局限且程度较轻,未见白色丘疹样损害。在很多DDEB患者,水疱随着时间的推移而减少,甲营养不良可能持续到成年。本病可致皮肤癌变,多年后发生基底细胞癌及鳞状细胞癌的均有报道。
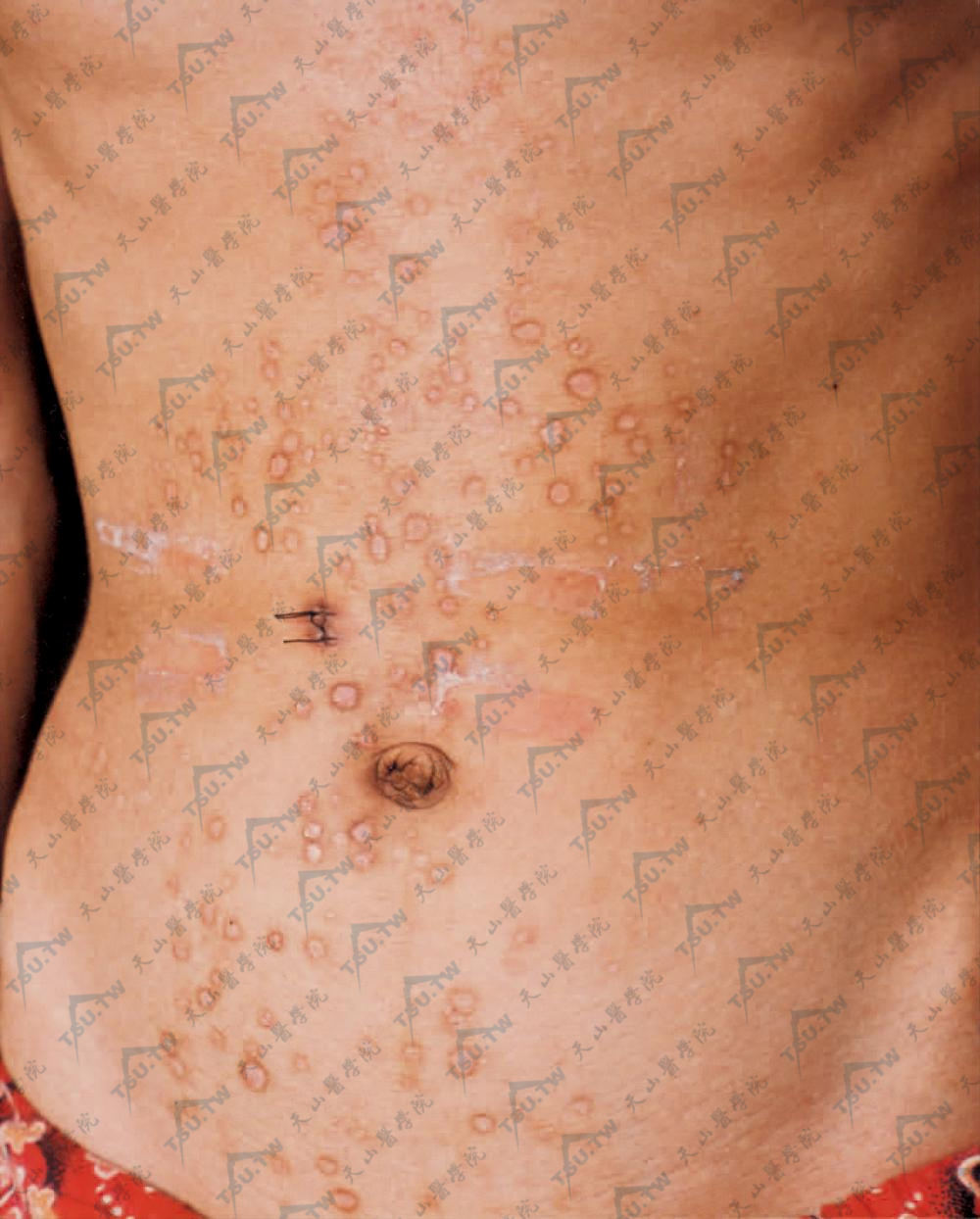
躯干见豆大至指甲大小坚实的象牙白色丘疹
二、隐性营养不良型大疱性表皮松解症(recessive dystrophic epidermolysis bullosa,Hallopeau-Siemens,RDEB)
RDEB有3种类型:泛发型、局限型和反相型。泛发型分为轻症(非Hallopeau-Siemens型RDEB,non-Hallopeau-Siemens RDEB,RDEB-nHS)和重症(Hall-opeau-Siemens型RDEB,Hallopeau-Siemens RDEB,RDEB-HS)。轻症主要表现为局限于手、足、肘和膝部的水疱。重症的特征是先天性泛发性皮肤和黏膜水疱。手指和脚趾被瘢痕包裹。
本病属常染色体隐性遗传,发病率约为1/30万。临床症状重于显性营养不良性大疱表皮松解症。损害见于刚出生或早婴儿期。出生时即有水疱和糜烂,粟丘疹、萎缩和瘢痕、贫血和生长迟滞同时存在。疱大而松弛,可为水疱和血疱。在皮肤的任何部位自动发生,尼氏征阳性。疱愈后通常留下萎缩性瘢痕,罕见情况下可留下大的糜烂面,形成皮肤缺陷,在四肢伸面尤为突出。手指和足趾可被瘢痕组织包裹,形成棒状手。手X线摄片可见骨骼结构正常,仅畸形范围及瘢痕内被固定,丧失功能。如果屈曲面瘢痕粘连,也易使肘、膝固定在非功能位上。在萎缩性瘢痕的部位,特别在小腿上,鳞状上皮癌可以发生,并且是死亡的主要原因。肿瘤是多发性的,侵袭性生长,可广泛转移。
黏膜损害经常存在,唇、口腔、舌、喉、鼻、气管、咽、食管、生殖器、肛周均可累及。大疱、糜烂和溃疡可使进食困难,瘢痕可以使龈唇沟消失,限制舌的活动,喉部可同样受累,但以食管的复发性大疱最为常见,也最为严重。食管病变可以发生在幼儿期,但在成人期明显。吞咽困难是最主要的症状,往往可导致吸入性肺炎。甲营养不良性变和指顶硬化。牙排列不规则。头发和毳毛稀疏,有时可伴斑秃。
本病严重病例可因大面积表皮剥脱致液体丧失、感染、败血症、肺炎或由于食管收缩致继发营养不良而死亡。
三、胫前营养不良型大疱性表皮松解症(pretibial dystrophic epidermolysis bullous,DDEB-pt)
及痒疹样营养不良型大疱性表皮松解症(dystrophic epidermolysis buttons pruriginosa,DDEB-Pr) 1946年Kuske报道1例发生于胫前区的瘙痒性水疱、萎缩及瘢痕的DEB,称为胫前DEB。1994年McGrath等报道一种特殊的DEB,其特点为有剧烈的瘙痒性紫红色丘疹或结节,轻度的水疱及糜烂,呈线状排列。主要发生在颈前、前臂,少数亦可发生于躯干(下3图)。可出生时即发生,亦可在婴儿期或儿童期发病。成人期损害可呈苔藓样斑块,可见瘢痕、粟丘疹及甲营养不良。血中IgE水平可增高。要与肥厚性扁平苔藓、皮肤淀粉样变及人为皮炎鉴别。
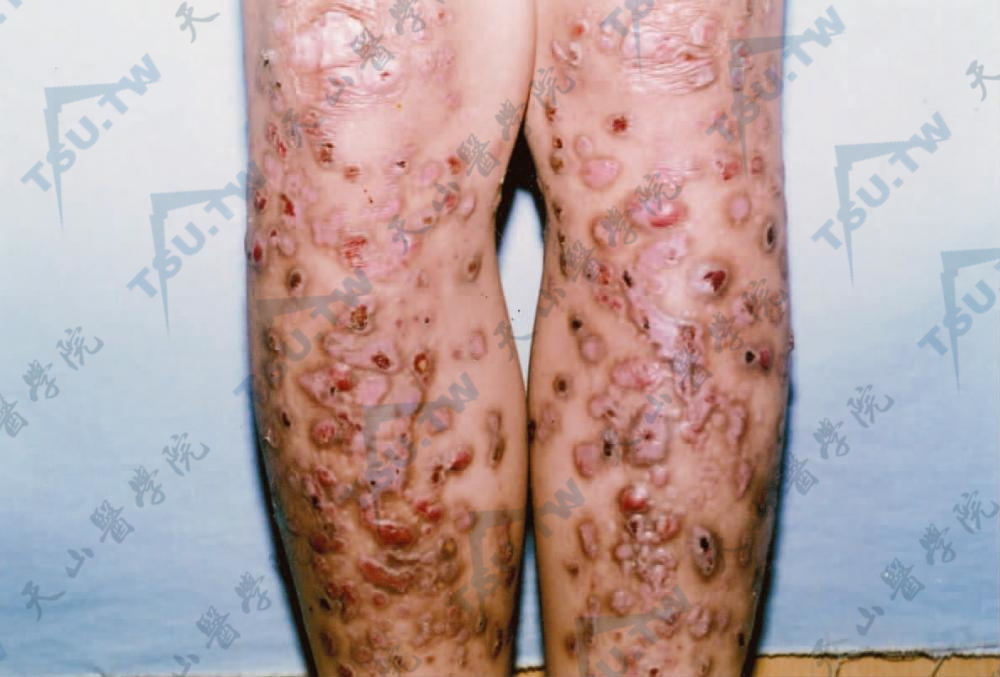
胫前散在圆形或椭圆形隆起呈暗棕红色的痒疹样结节,条状瘢痕,可见透明水疱(南阳市第一人民医院 翟立新提供)
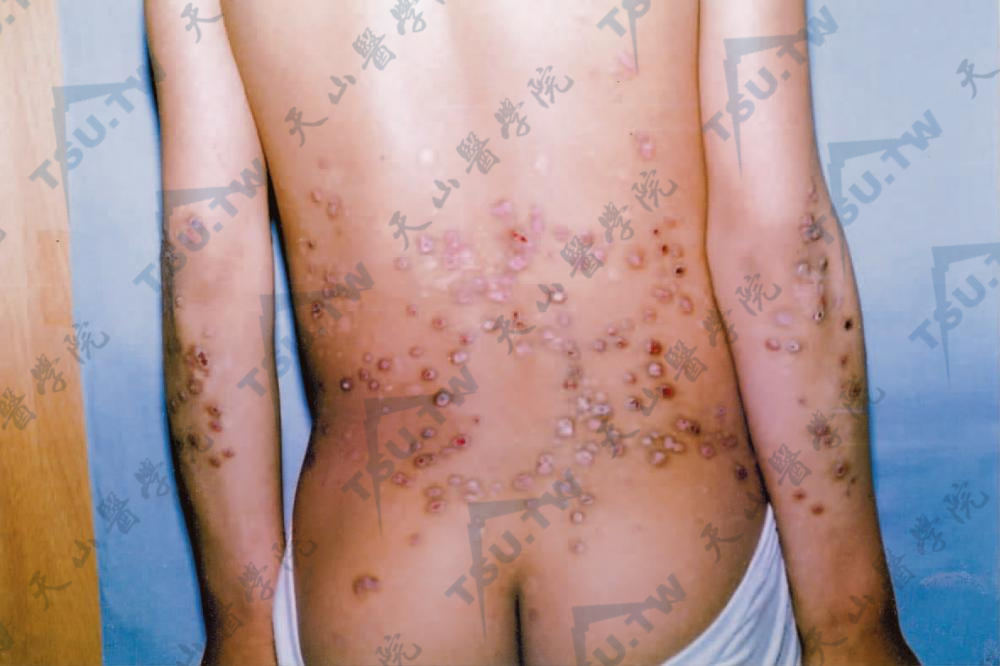
背部及上肢伸侧散在圆形、暗红色结节有血痂。见愈后瘢痕(南阳市第一人民医院 翟立新提供)
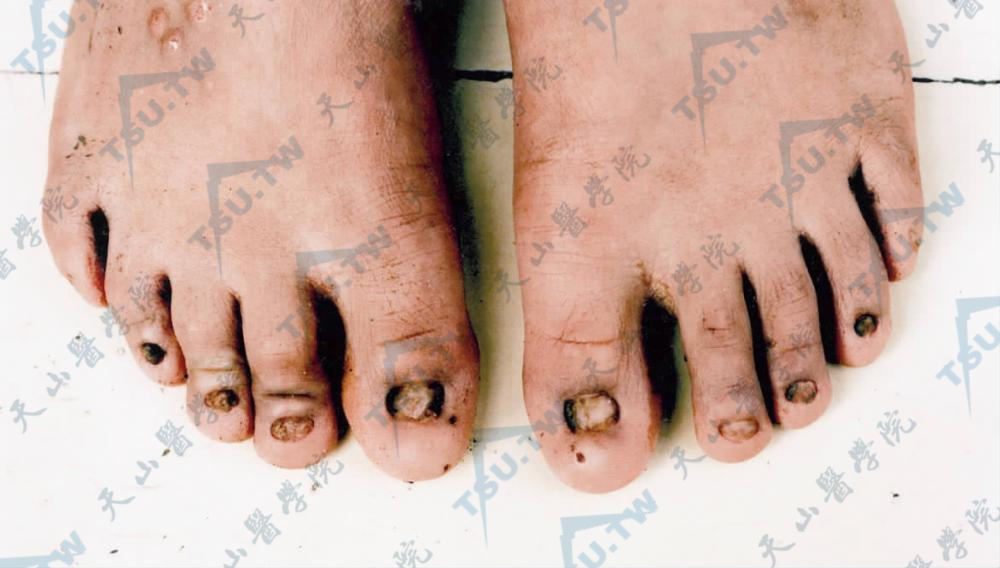
趾甲发育不良,甲板萎缩变形(南阳市第一人民医院 翟立新提供)
四、新生儿暂时性大疱性表皮松解症(transient bullous dermolysis of the newborn)
1985年,Hashimoto等报道了1例新生儿,在每一个轻微创伤处发展成水疱。分离位于基底板下,伴锚纤维和胶原蛋白变性。4个月后迅速愈合。甲不受损,且无瘢痕。目前认为该病的诊断标准是:
- 出生时或摩擦后出现水疱、大疱;
- 年龄数月时自愈;
- 无营养不良性瘢痕;
- 开始于真皮乳头内的表皮下水疱;
- 超微结构显示胶原蛋白溶解和锚纤维受损;
- 粗面内质网巨大性扩张,伴空泡内角质形成细胞的卫星小体形成。出生时或摩擦后出现水疱、大疱,一般在1岁时水疱消退,无瘢痕形成。
致病基因为编码Ⅶ型胶原蛋白的COL7A1基因颠换突变。
五、巴特综合征(Bart syndrome)
1966年Bart等首先报道本病,为常染色体显性遗传。小腿特征性先天性局限性皮肤缺损,四肢伸侧、间擦部位、颈部及臀部机械性大疱,口腔糜烂(出生时没有,开始喂食时发生),甲缺乏或畸形。皮肤及黏膜糜烂愈合后无明显瘢痕,偶伴色素减退。预后良好,出生后1年症状常可自行消退。血缘家族中可有单独发生口腔糜烂、甲畸形或复发性水疱者。目前认为该病是DDEB的一种临床变异型,由编码Ⅶ型胶原的COL7A1基因(染色体3p)缺陷所致。
六、肢端角化性皮肤异色病(acrokeratotic poikiloderma,Weary-Kindler)
又称金德勒综合征(Kindler syndrome),1954年Kindler报道了1例先天性皮肤异色症合并由微小创伤引起的足部创伤性水疱的病例。现在已报道的病例不足100例。此型亦被认为是DDEB的变异型。其特征包括肢端大疱、伴明显萎缩的全身皮肤异色症、光敏感和肢端角化。据报道,有1病例出现假性阿洪病和硬化带。牙龈红斑与糜烂和快速进展性牙周病也可发生。组织学上,主要特征是真皮乳头层内弹性纤维缺乏和真皮中间层分裂。
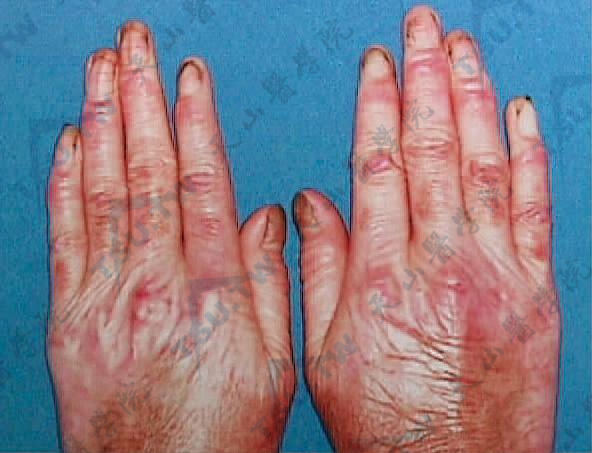
手背皮肤萎缩,伴有甲营养不良(第四军医大学西京医院 赵小东提供)
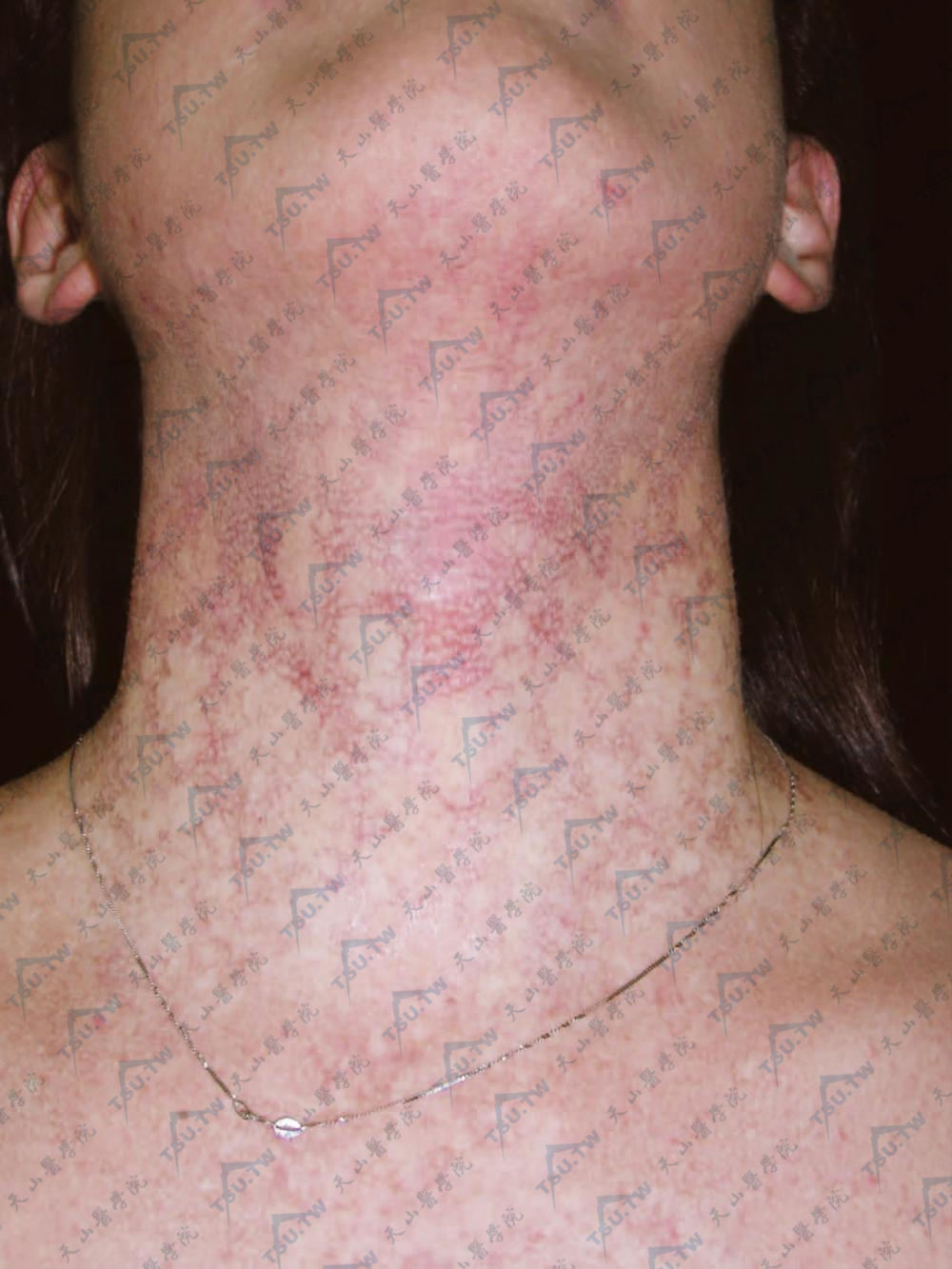
颈部和上胸部网状色素沉着和色素减退斑(北京协和医院 马东来提供)
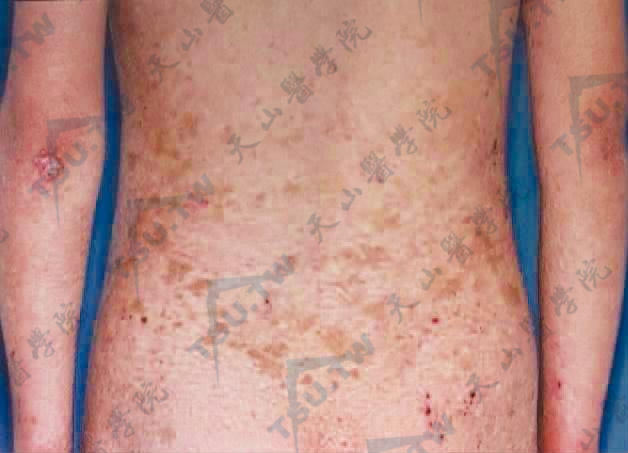
腰背部皮肤异色改变(第四军医大学西京医院 赵小东提供)
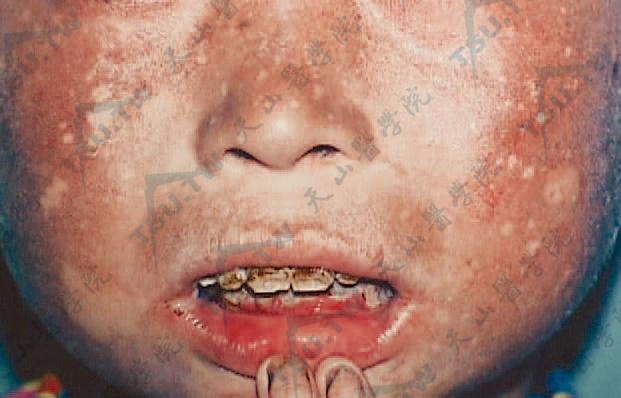
齿呈“四环素样齿”(第四军医大学西京医院 赵小东提供)
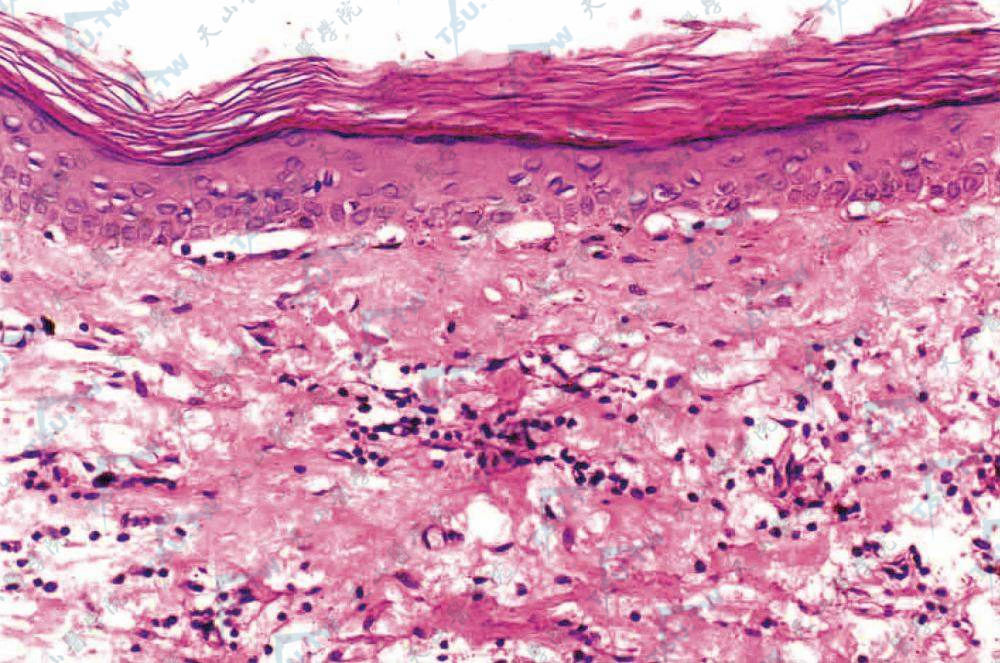
表皮萎缩,基底细胞部分液化变性,真表皮间可见有裂隙形成(HE染色×200)(第四军医大学西京医院 赵小东提供)
组织病理
单纯型大疱陈旧水疱分裂部可见于角层下。但在早期损害的基底细胞内可见有空泡形成及变性,其原始裂隙部位或在基底细胞层或由于基底层细胞完全分离而位于表皮下。在真皮乳头层中,血管扩张,无细胞浸润,弹性纤维正常,酸性磷酸酶和酸性黏多糖均正常。
交界型水疱发生在表皮和真皮间,通常不伴有大量的炎细胞浸润。有时基底细胞发生空泡化,有时则完全与真皮分离,基底膜见于大疱底部,可能系基底细胞与基底膜交界处被溶解酶所作用之故。
营养不良型大疱性表皮松解症与上述两型不同,其特点:表皮虽变薄且扁平,但基底细胞层正常,水疱分裂部位在PAS阳性基底层之下。乳头血管扩张,胶原在真皮上部减少而弹性纤维则增加,在真皮乳头部黏多糖曾加。
通常,光学显微镜下很难区别表皮内下部和表皮下水疱,不能鉴别透明板内型(如交界型EB)和致密板下型(如营养不良型EB)EB,需应用透射电子显微镜和特殊类型的免疫组织化学(即免疫荧光抗原定位)两种诊断方法。透射电子显微镜通过对皮肤水疱形成的超微结构变化,区别3种主要类型的EB,例如基底膜张力细丝、半桥粒、基底层下致密板、锚丝和锚纤维。抗原基因定位联合使用抗基底膜单克隆抗体,通过定性和半定量表达的特异性皮肤相关蛋白(特别是层粘连蛋白332,和Ⅶ型和X Ⅶ型胶原),对鉴别一些EB亚型有帮助(见上表2)。
诊断及鉴别
EB的诊断是比较容易确立的,但是仅根据家族史和临床表现确定遗传性EB的亚型很困难,需要鉴别的疾病有外胚层发育不良、肠病性肢端皮炎、色素失禁症、先天性局部皮肤缺损、自身免疫性大疱病(类天疱疮、天疱疮、线状IgA大疱性皮病、获得性EB)、一些感染性疾病(单纯疱疹、金黄色葡萄球菌烫伤样皮肤综合征、脓疱病)大疱性肥大细胞增生症等。
预防及治疗
目前,无特别有效的治疗方法。将来,对一些类型的EB或者EB亚型,基因治疗可能成为现实。已有一些初步成功的研究报道。
目前,处理EB的方法是防止机械性外伤和感染。局部外用抗感染药和口服抗生素阻止了继发的脓疱病。Weber-Cockayne EBS、多汗症通过局部应用水合氯化琥珀胆碱铝能够得到缓解,但其对水疱疗效不肯定。糜烂溃疡面应用各种人造敷料及组织培养的人造皮肤以加快皮肤愈合。但后者价格高且并不耐用。
系统使用苯妥英过去应用在营养不良型EB和交界型EB患者,但是疗效短暂,故在遗传性EB常规使用苯妥英的争议较大。经验表明,系统使用四环素对单纯型EB有临床疗效。沙立度胺能改善痒疹样营养不良型大疱性表皮松解症患者痒疹的症状。
系统使用糖皮质激素对任何类型的EB无效。曾有报道全身泛发的RDEB患者应用低剂量糖皮质激素有一定疗效。其他皮肤外并发症需对症处理。对慢性致残和毁形疾病需进行心理疏导。
