
又名表皮松解型类天疱疮(epidermolytic pemphigoid),本病是一种自身免疫性慢性大疱性皮病,血循环中有抗Ⅶ型胶原的IgG抗体,HLA-DR2发生率高。
病因:Ⅶ型胶原是本病的抗原,它存在于基底膜致密板及其下方的锚纤维内,抗原分子量为290 kD的糖蛋白和145 kD胶原体。它由角质形成细胞和成纤维细胞所产生。患者血清中含有抗基底膜Ⅶ型胶原抗体,它能与锚纤维相结合形成免疫复合物并激活补体,产生趋化因子和吸引中性粒细胞至基底膜,后者释放蛋白酶,导致表真皮分离形成水疱。自身抗体为IgG,在慢性机械性大疱形成中,IgG1和IgG4占多数。
症状表现
本病多见于成年人,儿童和老年人也可发病。疾病早期临床表现变化多样,最少有5种类型的临床表现:
(二)经典型:以肢端分布的非炎症水疱,愈后留有瘢痕和粟丘疹为特征。皮损好发于易受外伤和受压部位,如手足、肘膝关节伸侧面,在无炎症的皮肤上出现水疱、大疱,部分为出血性,发展成脱屑、结痂、糜烂,愈后留下萎缩性瘢痕及粟丘疹(图3-13-28),临床上类似皮肤迟发型卟啉症,但不具备后者的其他特征,如多毛、皮损多在光照部、硬皮病样改变及尿卟啉阳性等。有些病例有瘢痕性斑秃、甲营养不良和甲萎缩。1/3患者可伴有黏膜损害,少数患者有广泛的黏膜损害,口腔和食管黏膜被累及。
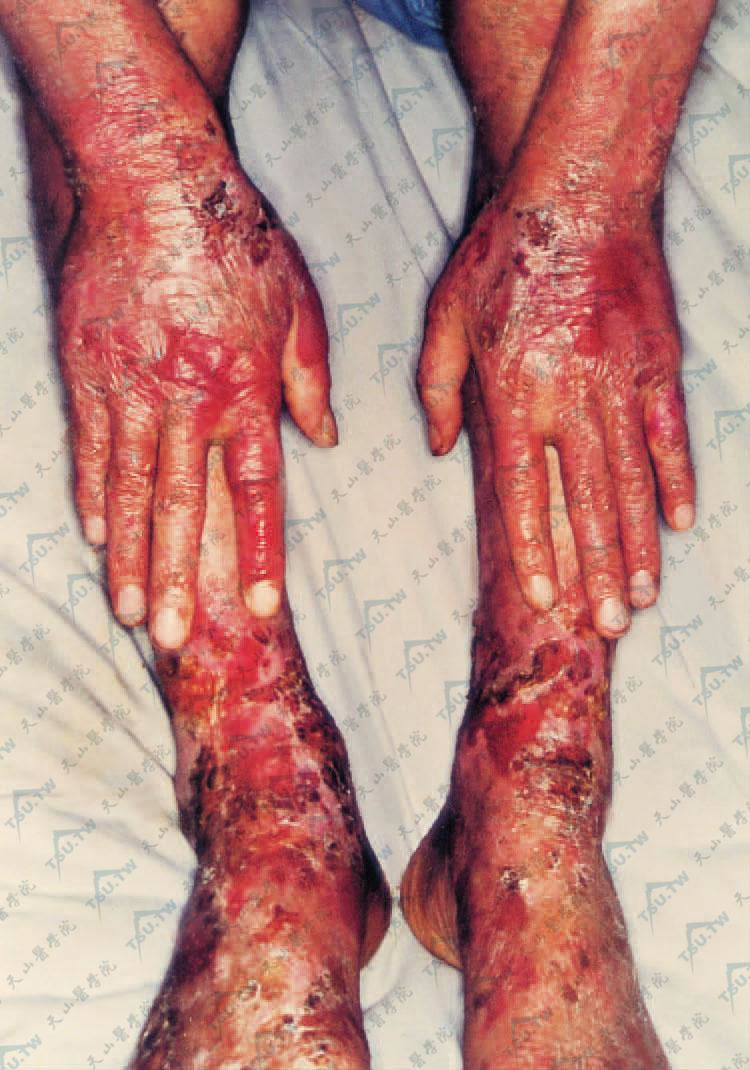
表皮松解症 四肢伸侧,水疱与血疱可见结痂与糜烂,愈后有轻度的萎缩性瘢痕,并常见粟丘疹及色素沉着
(二)大疱性类天疱疮型 累及四肢、躯干、皮肤褶皱处,为泛发性炎症性水疱、大疱,疱壁紧张,疱周炎症性红斑或水肿。也可仅为红斑或水肿性斑块而无水疱。患者常诉瘙痒,而机械性水疱、瘢痕、粟丘疹可不显著。
(三)瘢痕性类天疱疮型 部分获得性大疱性表皮松解症以黏膜受累为主,临床表现类似瘢痕性类天疱疮,在口腔、食管上端、结膜、肛门、阴道出现糜烂和瘢痕,而光滑皮肤上无损害。
(四)Brusting-Perry类天疱疮型 患者具有Brusting-Perry类天疱疮的特征,同时有针对Ⅶ型胶原的自身抗体。头颈部慢性复发性水疱大疱性皮损,留有瘢痕,很少累及黏膜。
(五)IgA大疱性皮病型 这一类型的获得性大疱性表皮松解症表现为表皮下大疱,嗜中性粒细胞浸润,直接免疫荧光可见基底膜带线状IgA沉积。此型表现类似IgA大疱性皮病、疱疹样皮炎、儿童慢性大疱性皮病。部分患者表现为紧张性水疱排列成环形,黏膜同时受累。自身抗体通常为IgA或IgG或两者均有。
20%~60%患者血清中可检测到Ⅶ型胶原的IgG抗体。67%~82%的患者HLA-DR2阳性。患者可伴有其他系统疾病,如肠炎、系统性红斑狼疮、淀粉样变、甲状腺炎、多发性内分泌腺病综合征、风湿关节炎、肺纤维化、慢性淋巴性白血病、胸腺瘤、糖尿病,及其他自身免疫性疾病。其中以肠炎最常见。
组织病理
为表皮下水疱,具体的病理表现因疾病的发展阶段不同,炎症性皮损病理显示大量的中性粒细胞为主的浸润。而机械性大疱无炎症阶段,则细胞浸润不明显。
直接免疫荧光:所有患者基底膜带均有线状IgG沉积,部分可见线状IgA和IgM。大部分有补体C3沉积。基底膜带染色比大疱性类天疱疮宽。用氯化钠分离表真皮,荧光沉积于真皮侧的基板及其下方。
间接免疫荧光:一半的患者血清中可检测到抗基底膜带的循环自身抗体(Ⅶ型胶原抗体)。
透射电镜:水疱位于基底膜致密板下方,锚纤维数目无减少。
诊断及鉴别
根据成人发病,易受外伤处发生水疱、瘢痕、粟丘疹,无大疱性表皮松解症家族史,直接免疫荧光检查基底膜带有IgG线状沉积,排除大疱性类天疱疮和大疱型系统性红斑狼疮。大疱性类天疱疮一般不形成瘢痕,水疱位于透明板内,抗原分子量为240 kD和180 kD,HLA-DR2阳性率不高,用氯化钠分离表真皮,IgG沉积在表皮侧。
治疗
糖皮质激素:本病对此治疗不敏感,仅适用于皮损广泛者,泼尼松每日1~1.25mg/kg,皮损控制后减量。国外报道糖皮质激素联合氨苯砜或氨苯磺胺可作为一线治疗,尤其对于儿童患者。
免疫抑制剂:硫唑嘌呤或甲氨蝶呤单独或与泼尼松联合应用可尝试。国外报道环孢素每日6mg/kg治疗有效。
大剂量免疫球蛋白静脉注射:个别病例报道有效。
