-
高脂蛋白血症(Hyperlipoproteinemia)
各种原因引起血浆脂质浓度升高并超出正常值范围称高脂血症(hyperlipidemia)。血浆脂质是以血浆脂蛋白的形式存在的,血脂升高的同时,其相应的脂蛋白浓度也升高,故又称高脂蛋白血症
1 -
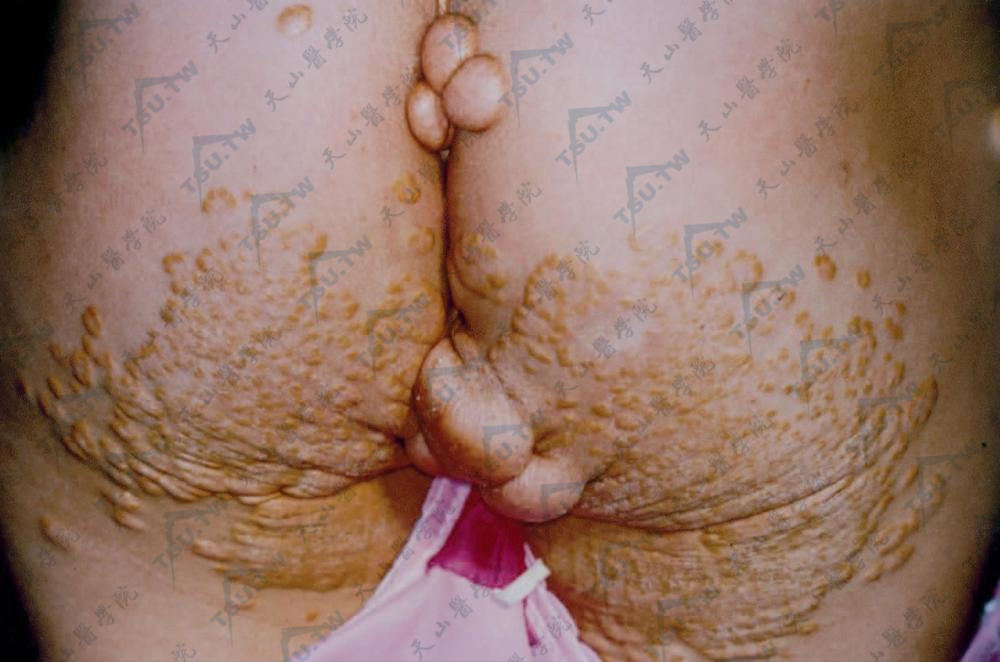
原发性高脂蛋白血症
一、家族性脂蛋白脂酶缺之(familial lipoprotein lipase deficiency)是一种罕见的常染色体隐性遗传性疾病,系由于LPL缺乏所致。LPL与CM和VLDL结合,使其中的甘油三酯水解为游离脂
2 -
继发性高脂蛋白血症
继发性高脂蛋白血症(Secondary Hyperlipoproteinemia):许多疾病或因素(如饮食、药物等)可产生高脂蛋白血症或使原有的高脂蛋白血症加重(下表),其中以糖尿病、甲状腺功能减退症、肾病
3 -
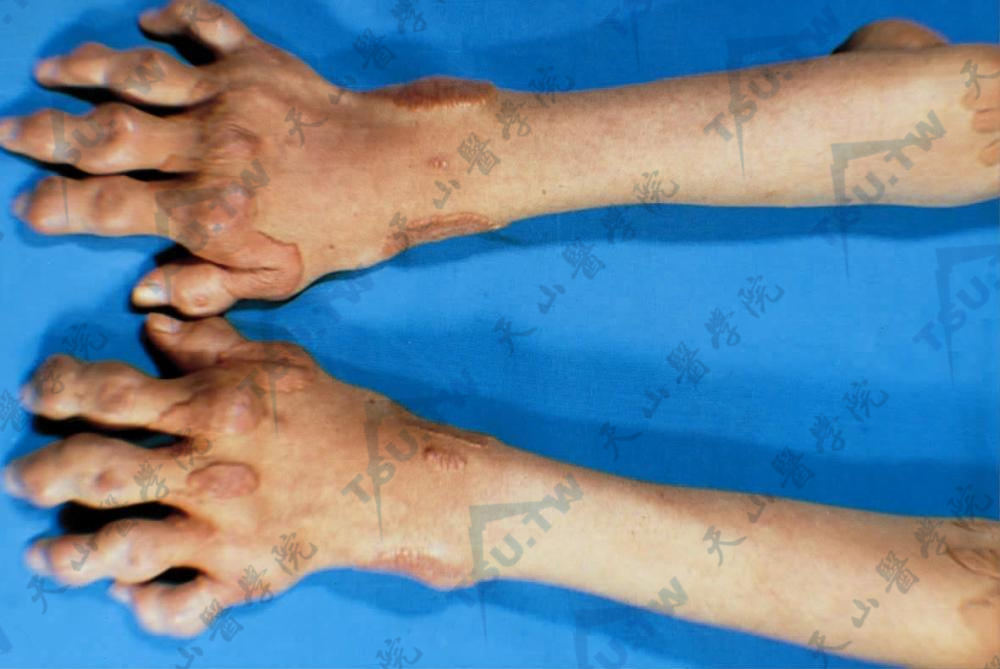
脑腱黄瘤病
本病又称胆甾烷醇病(cholestanolosis),是由于胆甾烷醇(cholestanol)和胆固醇广泛地沉积在眼晶状体、脑、肌腱及其他各组织内,并导致组织损伤和功能障碍的一种罕见的脂质代谢异常性
6 -
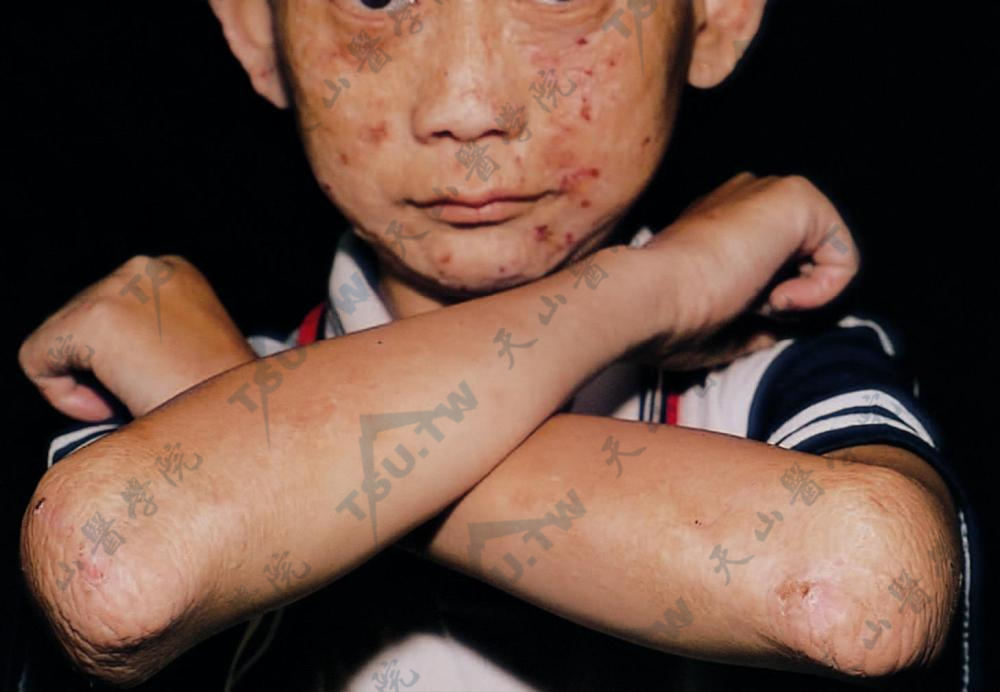
植物甾醇血症(Phytosterolemia)
本病也称谷甾醇血症(sitosterolemia)或假纯合子家族性高胆固醇血症(pseudohomozygous familial hypercholesterolemia),是一种少见的脂蛋白代谢障碍性疾病。其特征是黄瘤和血浆中
7 -
疣状黄瘤
疣状黄瘤(Verruciform Xanthoma)罕见,是一种疣样、乳头状或菜花样生长的良性增生性黄瘤样病变,好发于黏膜(偶尔见于皮肤)。1971年由Shafer首次报道。病因及发病机制迄今不明。有人
8 -
丹吉尔病
本病又名家族性α脂蛋白缺之(familial alphalipo-protein deficiency)或家族性局密度脂蛋白缺之(familialHDL deficiency)或β-脂蛋白缺之病(β-lipoprotein defici
9 -
尼曼-皮克病(神经鞘磷脂积累病)
尼曼-皮克病(Niemann-Pick disease)简称NPD,又称神经磷脂单核巨噬细胞病或神经鞘磷脂积累病(sphingolipidosis),是一种罕见的溶酶体贮积病。本病系(神经)鞘磷脂、胆固醇磷脂大量地贮
10 -
戈谢病(葡萄糖脑苷脂病)
本病又称葡萄糖脑苷脂病(glucosylcerebrosidosis),脑苷脂单核巨噬细胞病、脑苷脂贮积病(cerebrosidosis)、葡萄糖脑苷脂贮积病(glucosylceramide lipidosis)。本病是最常见的溶酶体
11 -
播散性脂质肉芽肿病(Farber病)
本病又称Farber病(Farber's disease)、法伯综合征(Farber's syndrome)、纤维细胞黏多糖障碍病(fibrocytic dysmucopolysaccharidosis)、脂质肉芽肿病(lipogranulomatosis)和Farber脂
12 -
类脂蛋白沉积症(皮肤黏膜透明变性)
本病又称皮肤黏膜透明变性(hyalinosis cutis etmucosae)和Urbach-Wiethe病,是指透明蛋白样物质沉积在皮肤、黏膜(口腔、咽喉)及内脏而引起的一种疾病。本病罕见,到2002年,世界共报
13 -
中性脂质贮积病(甘油三酯贮积病)
本病又名甘油三酯贮积病(triglyceride storage disease),是一种罕见的常染色体隐性遗传的脂质代谢障碍性疾病。Rozenszajn等于1966年首次报道2例常染色体隐性遗传性鱼鳞病患者
14 -
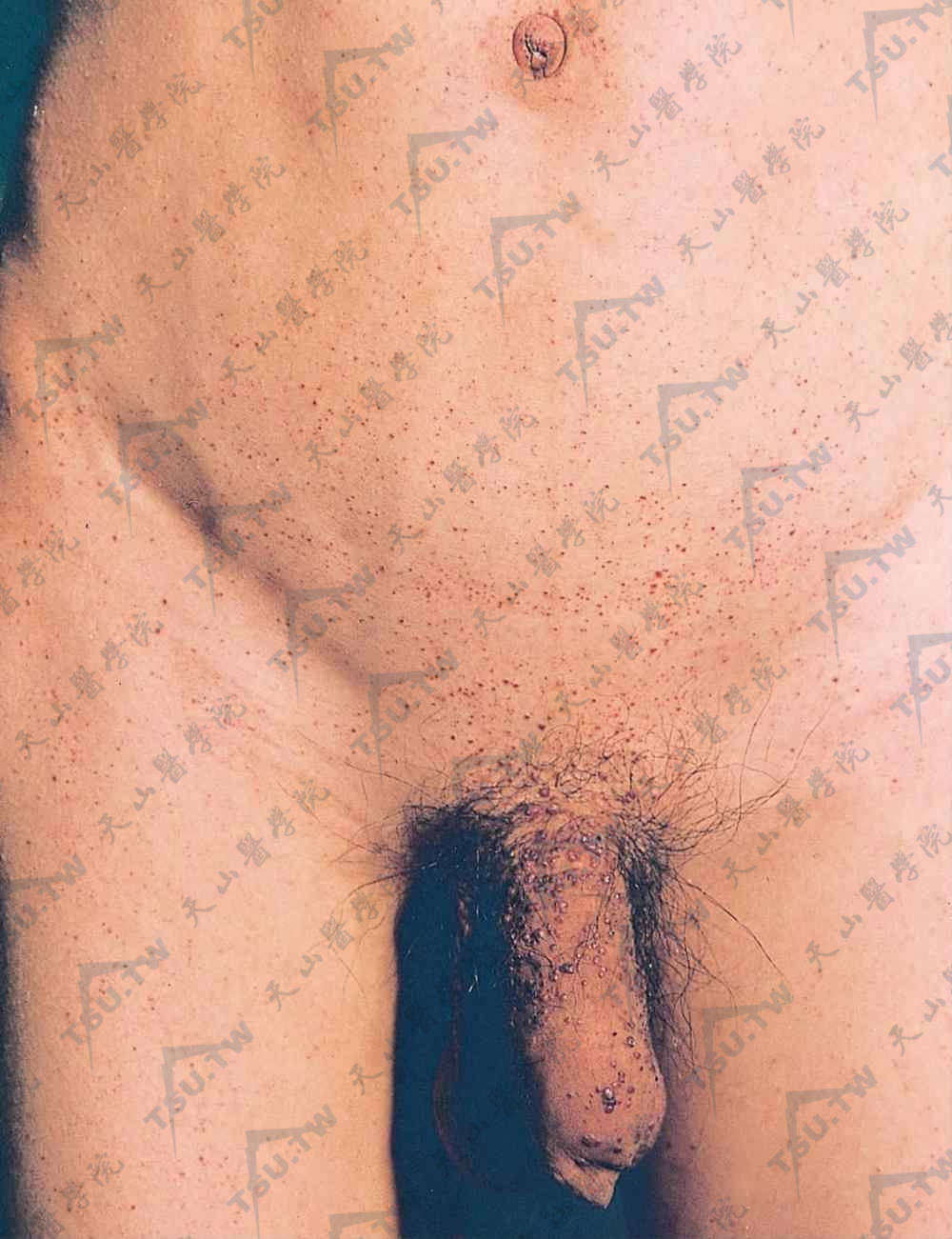
弥漫性躯体性血管角化瘤
弥漫性躯体性血管角化瘤(Angiokeratoma Corporis Diffusum、简称ACD)长期以来曾是Anderson-Fabry病的同义名,目前已知道它是几种少见的先天性溶酶体贮积病共同的皮肤标志,这些疾
15