-
原发性免疫缺陷病的基因定位
因遗传因素如染色体异常或基因突变所致的免疫缺陷病称为原发性免疫缺陷病(primary immunodeficiency disease,PID)。原发性免疫缺陷病的病因尚未完全清楚,但普遍认为与遗传有关
2 -
免疫缺陷病的常见皮肤表现
皮肤症状是免疫缺陷患者常有的临床表现,由于免疫缺陷的原因不同,其临床表现亦有差异,总结起来可有下列一些表现:(一)脓皮病:是免疫缺陷病最常见的皮肤症状之一。患者经常反复发生皮
3 -
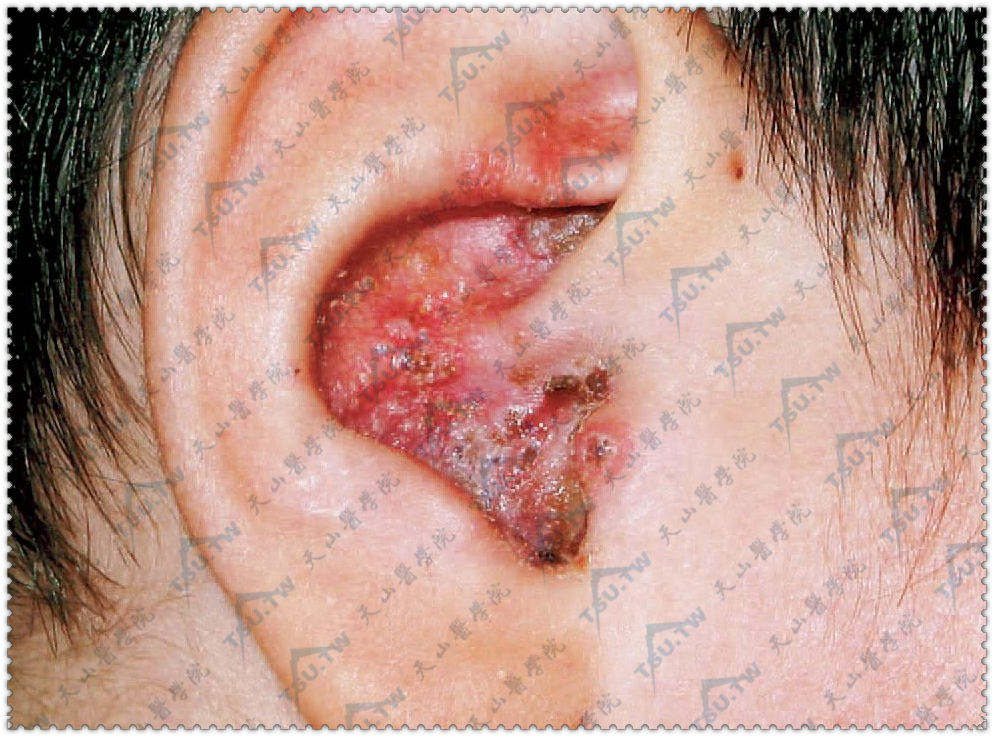
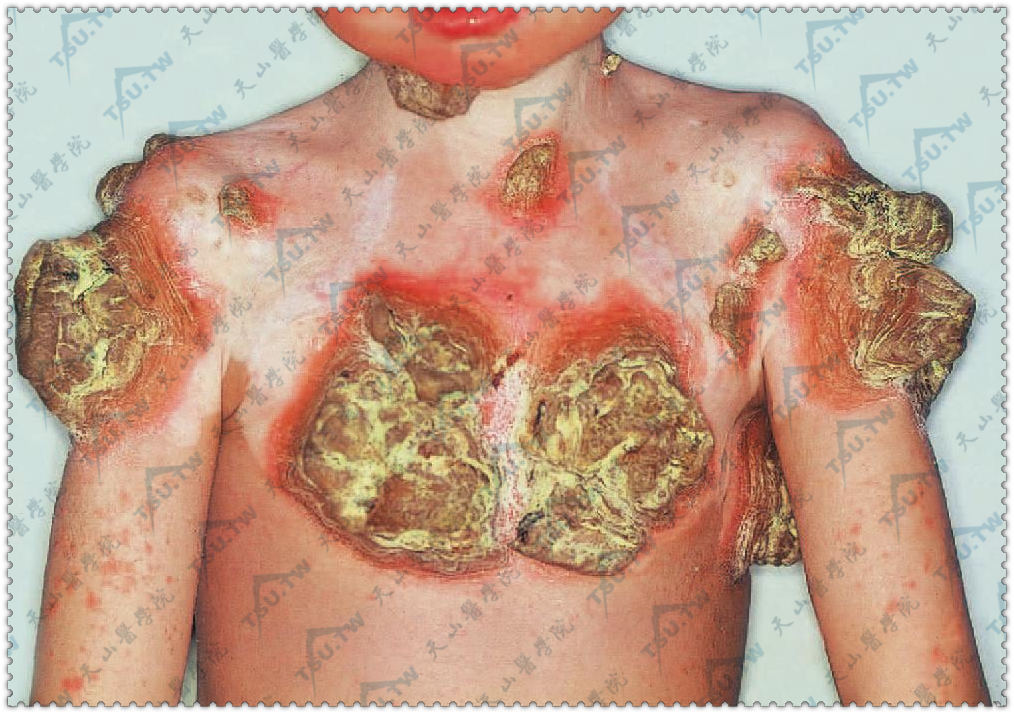
慢性黏膜皮肤念珠菌病
慢性皮慢性皮肤黏膜念珠菌病(chronic mucocutaneous candidiasis,CMC)是一种少见的慢性进行性念珠菌感染,临床表现为一组综合征,特点为慢性反复性的皮肤、指甲及黏膜的念珠菌感染
8 -
先天性无丙种球蛋白血症
先天性无丙种球蛋白血症(congenital agammaglobulinemia)属原发性免疫缺陷病,是由于B淋巴细胞发育障碍所致,大多数为X连锁无丙种球蛋白血症(X-linked agammaglo bulinemia,XLA),又称
9 -
特发性迟发性免疫球蛋白缺乏症
多数起病于10~30岁,男女发病数相等。最常见的临床表现为反复发作的鼻窦炎、支气管炎、中耳炎、肺炎。病原体为流感杆菌、肺炎球菌、溶血性链球菌、金黄色葡萄球菌等。皮肤表现
10 -
选择性IgA缺乏症
选择性IgA缺乏症(selective IgA deficiency)是指体液IgA含量极低,而其他免疫球蛋白含量正常或升高的一种体液免疫缺陷病。本症首由West等于1962年报道,是最多见的一种原发性体液
11 -
普通可变型免疫缺陷病
普通变异性免疫缺陷病(common variable immunodeficiency,CVID)又名获得性低丙种球蛋白血症,是一组发病机制和临床表现不均质的原发性免疫缺陷病,以严重的抗体产生障碍,通常以一种
12 -
单纯性原发性IgM缺陷症
此类病人血中IgM缺乏,其他类型免疫球蛋白正常。同种血细胞凝集素缺乏。患者易反复发生细菌感染,特别容易感染脑膜炎双球菌、肺炎双球菌及流感杆菌。多数患者易患寻常疣,1/5此类
13 -
高免疫球蛋白M综合征
高免疫球蛋白M综合征(hyper IgM syndrome)在20世纪60年代由Asselain和Rosen等首次报道。HIGM是一种罕见的原发性免疫缺陷病,主要表现为反复感染,血清IgG和IgA水平低下或测不出,而
14 -
重症联合型免疫缺陷病
本病为常染色体隐性遗传病,出生后1~2月内即开始发病,常在2岁内死亡。患儿不能自己合成免疫球蛋白,出生6个月后血清免疫球蛋白总量常低于250mg/L。外周血淋巴细胞计数常低于1.5&t
15