-
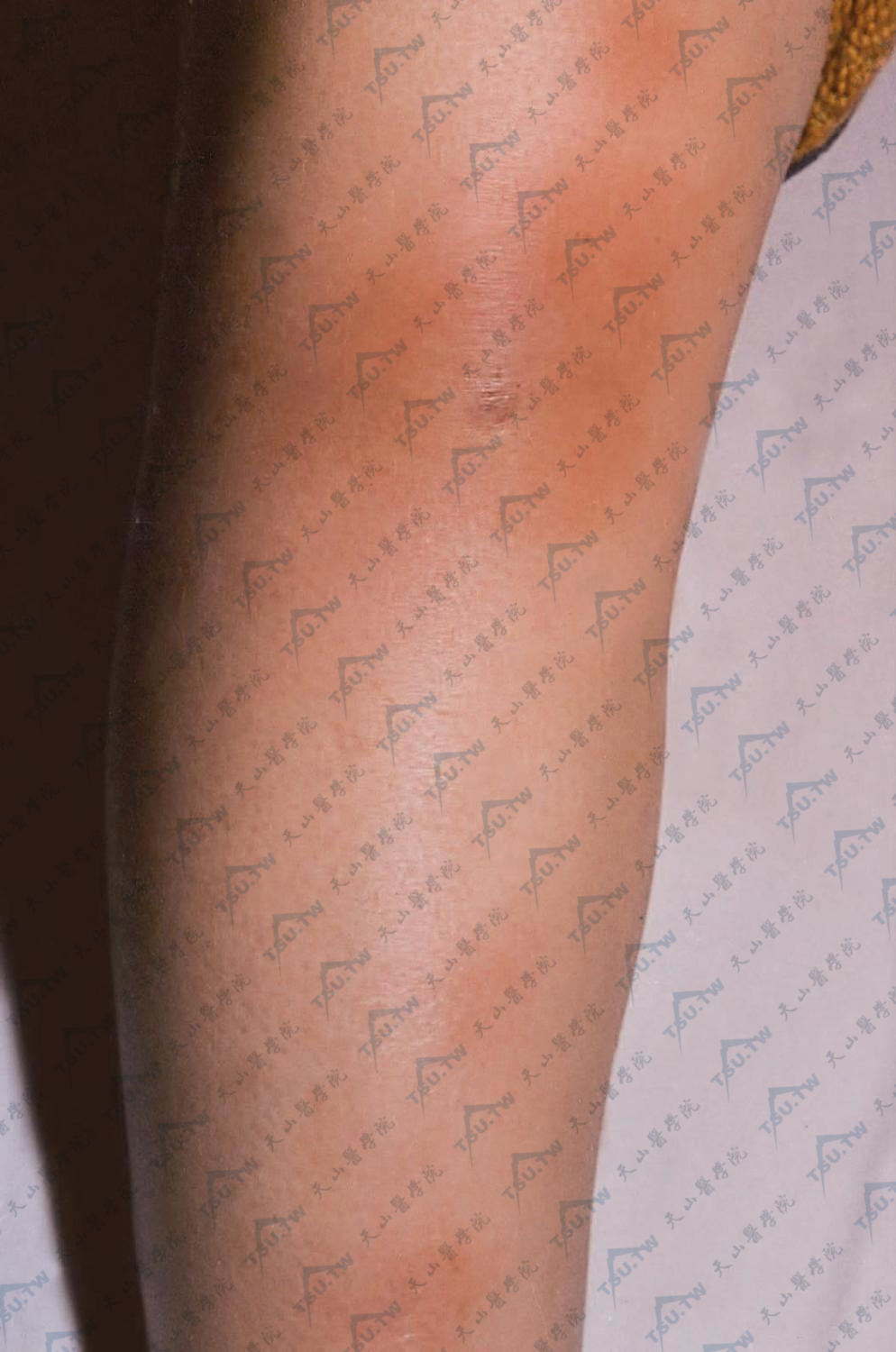
结节性红斑
结节性红斑是常见的炎症性脂膜炎,该病由Willan于1798年首次提出,1842年wilson认为结节性红斑是多形红斑的一型,1866年Hebra进一步描述其临床表现称挫伤性皮炎(dermatitis contus
2 -
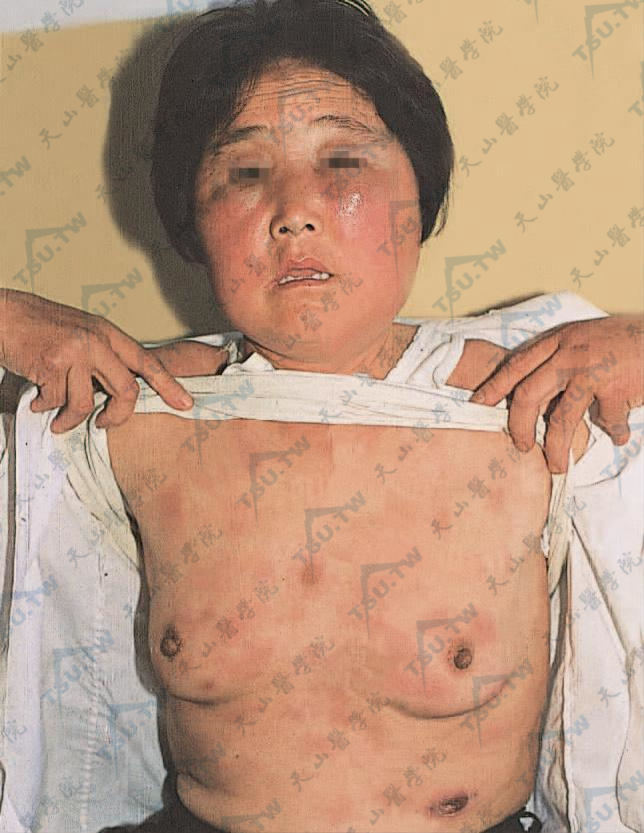
复发性发热性结节性脂膜炎(韦伯-克里斯汀病)
本病又名韦伯-克里斯汀综合征(Weber-Christian syndrome)、韦伯-克里斯汀病(Weber-Christian disease)、特发性小叶脂膜炎(idiopathic lobular panniculitis)、结节性脂膜炎(nodula
3 -
皮下脂肪肉芽肿病
本病又称罗思曼-马凯综合征(Rothmann-Makai syndrome)、罗思曼-马凯皮下脂肪肉芽肿(Rothmann-Makai lipogranulomatosis subcutanea)、罗思曼-马凯脂膜炎(panniculitis Rothmann-
4 -
胰腺性脂膜炎
本病由Chiai(1883年)首先描述,是一种合并胰腺疾患的脂肪坏死性炎症,除侵犯皮下脂肪外,可侵犯胰腺周围、腹膜和大网膜脂肪组织,引起内脏脂肪坏死。本病又称酶脂膜炎(enzymic pannicu
6 -
α1-抗胰蛋白酶缺陷性脂膜炎
自1972年以来,有很多复发性特发性有破溃的小叶脂膜炎与α1-抗胰蛋白酶缺陷有关。本病类似Weber-Christian综合征,主要与α1-胰蛋白酶抑制剂(α1-抗胰蛋白酶)缺乏
7 -
结晶储存性组织细胞增生症
结晶储存性组织细胞增生症少见,多数为个例报道。Kaufmman(1996年)报道1例61岁妇女皮下脂肪层有广泛的结晶储存性组织细胞浸润,患者伴有淋巴浆细胞性淋巴瘤,免疫表型为IgM kappa型
8 -
糖皮质激素后脂膜炎
本病也称类固醇后小叶脂膜炎(poststeroid lobular panniculitis),临床少见,所有报道病例均为儿童。Smith和Good(1956年)报道患有风湿热、白血病和肾炎儿童应用泼尼松治疗过程中,由
9 -
新生儿水肿
本病又名水肿性硬化症(edematous sclerema)。本病虽非皮下脂肪疾病,但临床症状与新生儿硬化症相似,亦发生于早产儿和营养不良婴儿,死亡率高。病因及发病机制:病因不明,可能与寒冷、
11 -
新生儿皮下脂肪坏死
Cruse(1933年)首先报道本病。本病发生于出生后4周内,常见于健康足月或过月产新生儿。组织病理为小叶性脂膜炎,有皮下脂肪细胞变性坏死和肉芽肿性炎症,形成皮下结节,可自行消退,很少
12 -
结节性囊性脂肪坏死
本病又名包裹性脂肪坏死(encapsulated fat necro-sis),是一种包裹性皮下脂肪坏死,表现为单个或多个皮下结节,30%的病例有外伤史。病因及发病机制早期损害表现为脂肪组织小叶被周围
13 -
脂肪膜性脂肪坏死
本病又名膜囊性脂肪坏死(membranocystic fat necrosis)、Nasu-Hakola病,是一种遗传性疾病,有长骨异常和系统性深在脂肪组织膜囊变性(membranocystic degeneration)以及有进行性嗜
14 -
寒冷性脂膜炎
本病又名E Frigore脂肪坏死(adiponecrosis E Frigore),由寒冷直接损伤局部皮下脂肪组织,引起局限性脂膜炎。Haxthausen(1941年)报道5例,国内王振国(1983年)报道1例为30天婴儿伴有支气
15