
本病较少见,为多系统疾病。具有吞噬能力的组织细胞增生引起的脂肪小叶性脂膜炎。临床上有泛发性红斑、疼痛性皮下结节,组织学上有组织细胞增生并吞噬红细胞、白细胞、血小板等形成“豆袋状细胞”。除皮下脂膜外,可侵犯肝、脾、浆液膜等。有发热、全血细胞减少、血甘油三酯升高和多脏器出血,可出现噬血细胞综合征(haemophagocytic syndrome,HPS),以往本病列入Weber-Christian综合征。1980年Winkelmann报道5例,均为女性,其中3例为青年,病程3~6个月不等,认为本病不同于Weber-Christian综合征,是一独立疾病。国内马圣清(1987年)、葛以信(1988年)、包为政(1996年)等均有病例报道。
病因及发病机制
本病为一种谱系疾病,目前尚不清楚本病性质,为原发组织细胞性还是原发T细胞性疾病。免疫组化研究发现皮损中组织细胞分化良好,推测本病为组织细胞对潜在良性或恶性T细胞增生的反应。有的病例为病毒感染(如EB病毒、巨细胞病毒、HIV)触发。
该病凝血功能障碍的机制还不清楚,可能为血小板减少、凝血因子Ⅷ减少、纤维蛋白原水平降低和纤维蛋白降解增多等综合因素造成,或增生的淋巴细胞释放蛋白水解酶进入血液所致,而非凝血酶异常。有报道骨髓同种异体移植后可出现组织细胞吞噬性脂膜炎。
临床症状
急性或慢性发病,可反复发热、体重减轻,皮损为多发的炎症性皮下结节和斑块,红色有触痛,直径可达2~20cm,可发生于身体各处,好发四肢,偶见于躯干及面部。结节可发展为紫癜性或青肿样外观,自然破溃。可侵犯口腔、阴道等处黏膜形成多数溃疡。有肝、脾、淋巴结肿大,进行性肝功能减退,出现黄疸、凝血机制障碍、浆膜炎以及肾衰竭。可死于肝、肾衰竭、肺炎和胃肠、泌尿、呼吸道出血。亦可发展为T细胞淋巴瘤、B细胞淋巴瘤、组织细胞性淋巴瘤或窦性组织细胞增生症伴巨大淋巴结病。
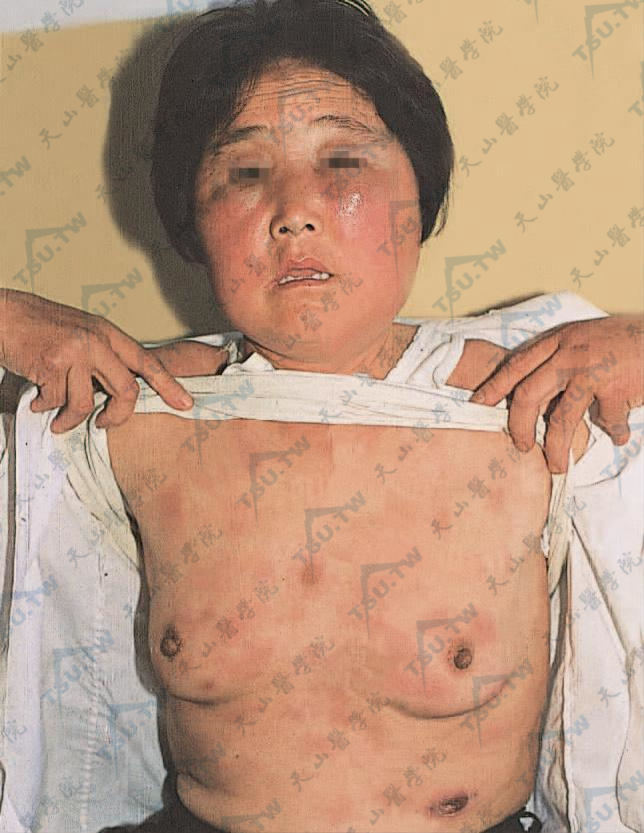
面、胸部多数鲜红色斑及皮下结节,大小不一,直径可达2~20cm
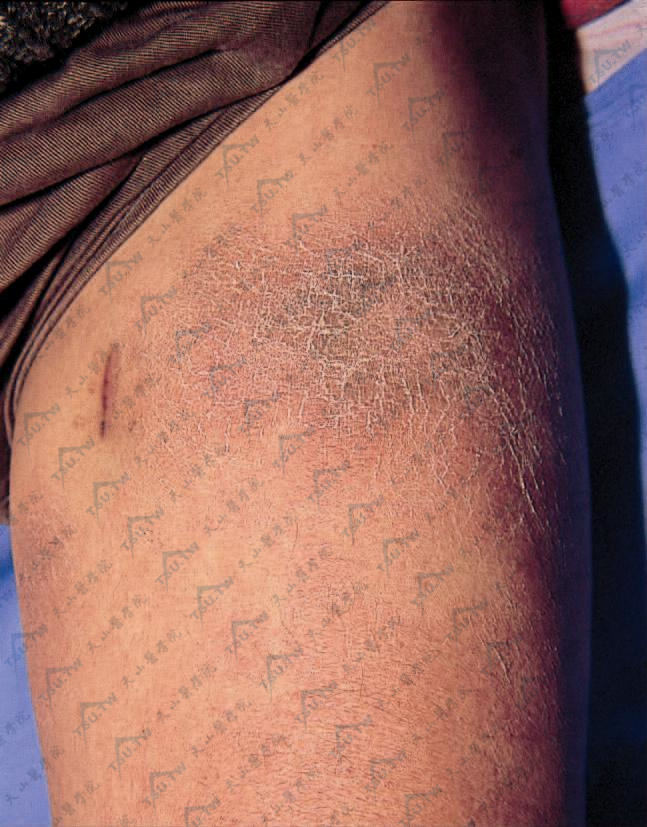
大片鲜红色斑块及皮下结节
实验室检查 白细胞、血小板减少,贫血,血浆白蛋白降低,低血钙,γ球蛋白升高,类风湿因子阳性,AST升高,γGTP、碱性磷酸酶、淀粉酶和脂酶值均升高,纤维蛋白原减少,纤维蛋白分解产物增加以及凝血机制异常。
组织病理
为小叶型脂膜炎,有灶性脂肪坏死和组织细胞增生。少数病例初期为间隔性炎症。大部分病例累及骨髓、淋巴结等单核吞噬细胞系统。受累组织逐渐被合胞(syncytium)组织细胞取代,可伴有T淋巴细胞(主要为辅助性T细胞)及浆细胞浸润,有红细胞外溢,在炎症边缘区可见组织细胞吞噬淋巴细胞、中性粒细胞、红细胞、血小板及其细胞碎片,形成特征性的“豆袋状细胞”(图3-23-5,图3-23-6)。免疫组化检查发现组织细胞为良性,淋巴样细胞成分中主要由T细胞组成。骨髓、肝、脾和淋巴结被累及时也可见到相似的浸润。
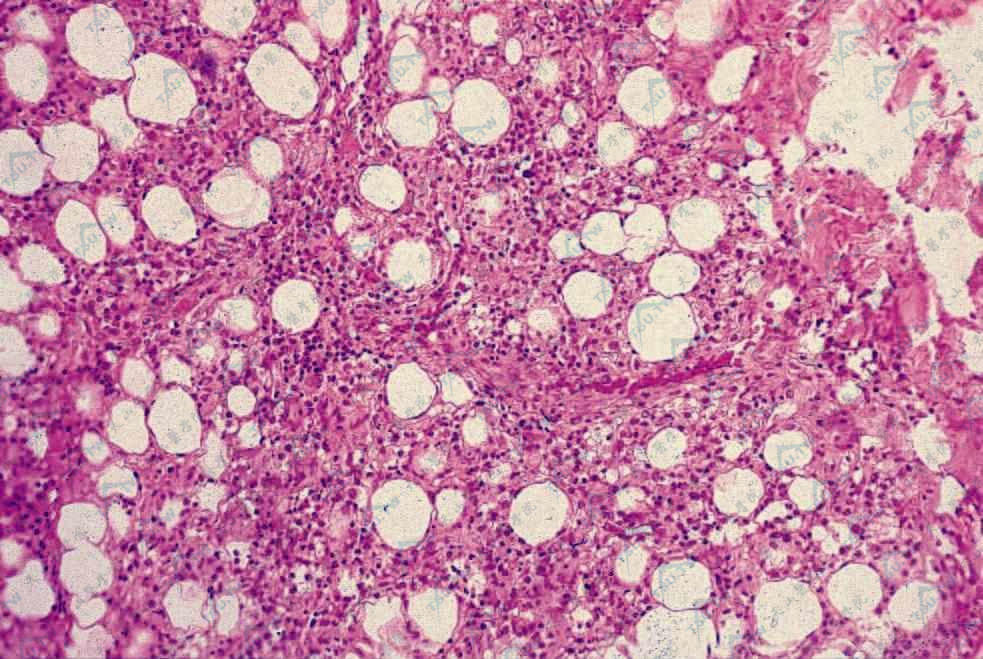
脂肪小叶间隔弥漫性炎症(HE染色×100)
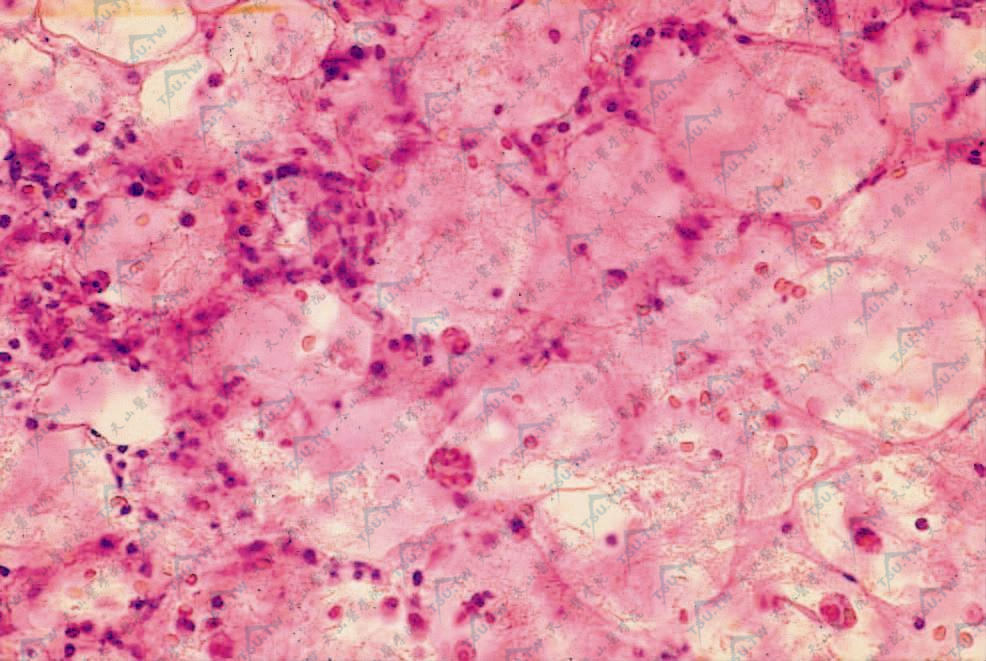
右下炎症浸润中见“豆袋状细胞”(HE染色×400)
诊断、治疗
本病反复发热,有皮下脂膜炎,肝、肾功能损害,浆膜炎和出血倾向等,组织病理变化有“豆袋状细胞”,可与Weber-Christian病鉴别。其次要与恶性组织细胞增生症鉴别,后者增生的组织细胞有异型性,临床症状更严重,病程短,一般3~6个月死亡。
尚无有效治疗方法。如无恶性病变,可用环孢素治疗,有恶性病变时进行化疗,进行期病例用联合化疗可延长生存期,甚至完全缓解。化疗药物包括泼尼松、环磷酰胺、苯丁酸氮芥、博来霉素、长春新碱、多柔比星。糖皮质激素和硫唑嘌呤联合治疗或静脉滴注丙种球蛋白治疗有效。
本病死亡率高,常因累及骨髓和肝脏等发生内脏出血死亡。但本病也可自然缓解,或经治疗后可以长期缓解。
