
本病又称黑利-黑利病(Hailey-Hailey disease),是一种遗传性皮肤病,其临床特征是在颈、腋、腹股沟反复出现水疱、糜烂,尼氏征阳性。无全身症状,慢性经过。无性别和种族的差异。
病因及发病机制
本病是一常染色体显性遗传性皮肤病,70%的病人有家族史,致病基因定位于3q21-22,与编码一种新型钙离子泵的基因ATP2C1基因的多个突变有关。在培养的角质形成细胞中,钙的调节受损。这种钙离子泵在维持高尔基体的钙离子浓度中起重要作用,ATP2C1基因突变导致角质形成细胞间黏附障碍,外界刺激如摩擦、寒冷、紫外线照射后最终发生棘刺松解,裂隙形成。葡萄球菌、酵母菌和病毒感染会使患者皮损加重。
用患者角质形成细胞或表皮作器官培养,可以发生棘刺松解,棘刺松解能被糖皮质激素和丝氨酸蛋白酶所抑制。血纤维蛋白溶酶原可以引起棘刺松解或使其明显。
临床症状
本病多在青春期发病,好发于颈、项部、腋窝和腹股沟,少见于肛周、乳房下、腘窝和躯干等部位,病变可局限于上述一两处,也可泛发。少数患者可有黏膜损害,主要累及口腔、喉、食管、外阴及阴道。基本损害是成群小疱或大疱在外观正常皮肤上或红斑上发生。疱液早期澄清,很快混浊,破裂后留下糜烂面和结痂,中心渐愈,周边又出现新皮疹,而呈环形,也可呈扁平柔软、湿润增殖面,常有瘙痒,并伴有腥臭。有时自觉疼痛,特别是发生裂隙时。水疱尼氏征阳性,也可以阴性。不典型损害有斑丘疹、角化性丘疹、乳头瘤样增殖。常见由白念珠菌、疱疹病毒和金黄色葡萄球菌引起的继发感染。
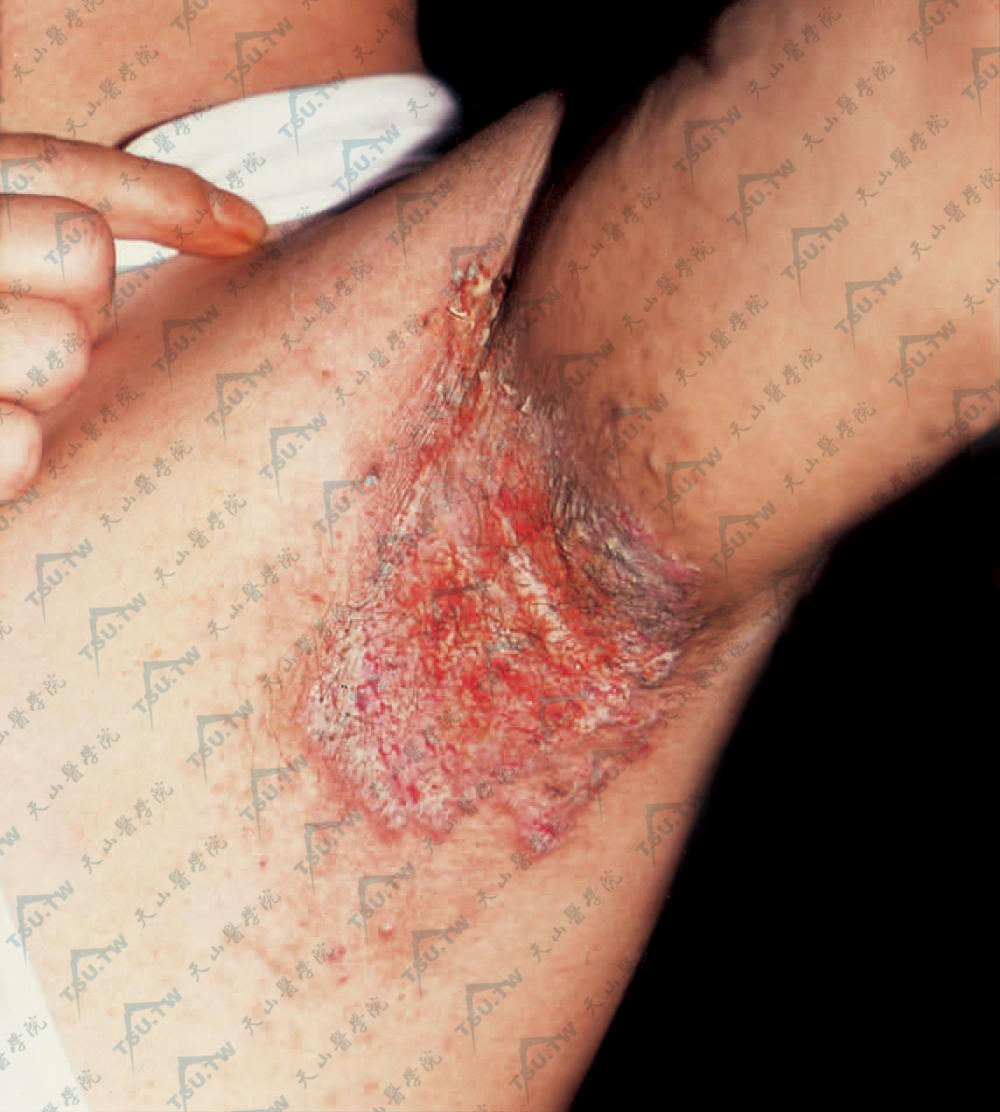
腋窝有成群的小疱或大疱,破后有糜烂或结厚痂,周边仍有新疹发生,使皮损呈环形、湿润的增殖面
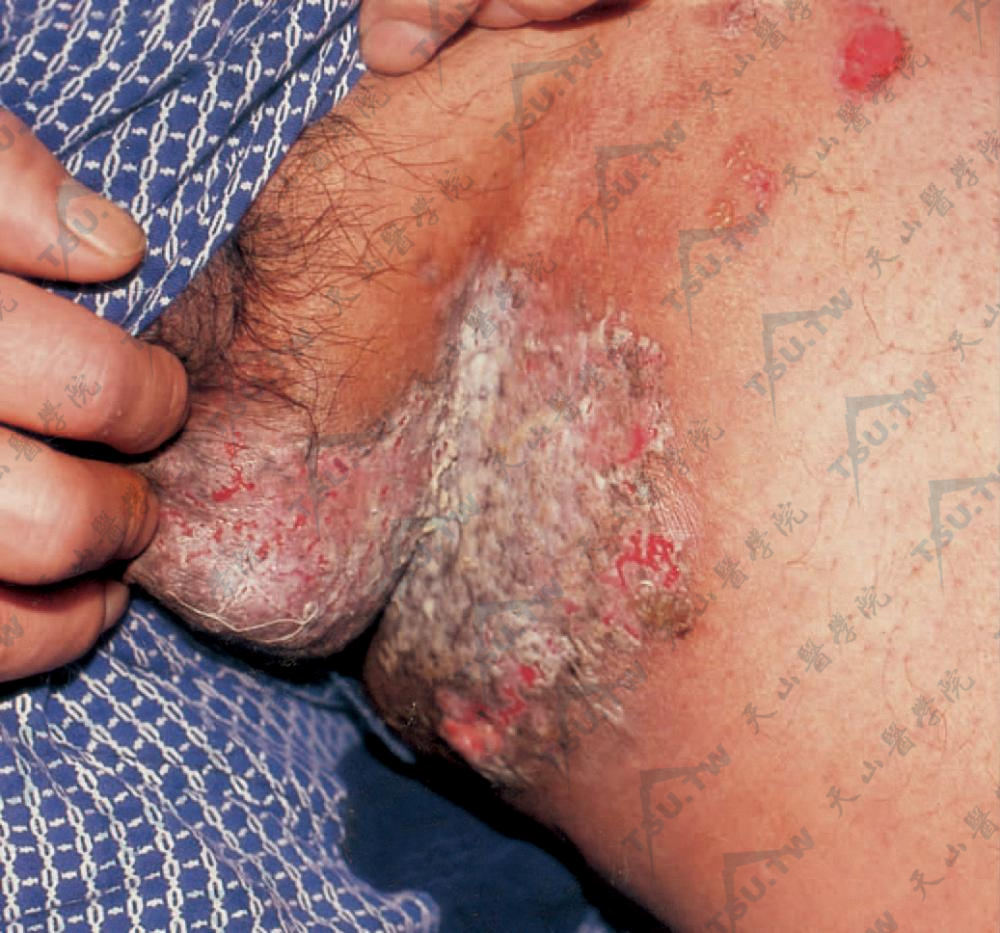
阴股部见红斑、糜烂、结痂,呈湿润增殖面
掌跖偶有点状小坑及散在分布的角化。约70%的患者指甲有白色条纹。少数患者有黏膜损害,有时在肛门生殖器部位可出现多个直径3~5mm的疣状丘疹,类似于尖锐湿疣。病变附近淋巴结可以肿大或疼痛。
损害的发生与机械性外伤、压力和紫外线照射相关。冬天症状常减轻甚至消失,夏天则趋于加重。皮疹经过数周可自行消退,以后又往往在原处复发。
组织病理
基底层上裂隙形成和大部分表皮内出现部分性或完全性棘刺松解为本病的特征。后者呈塌砖墙样(dilapidated brick wall)外观。较成熟损害内有水疱和大疱形成,衬以单层基底细胞的乳突(绒毛)向上突入水疱腔或裂隙内。直接免疫荧光检查阴性。电子显微镜检查示张力细丝与桥粒分离,核周电子致密物聚集,角质形成细胞周围有许多延长和分枝的微绒毛,桥粒减少。
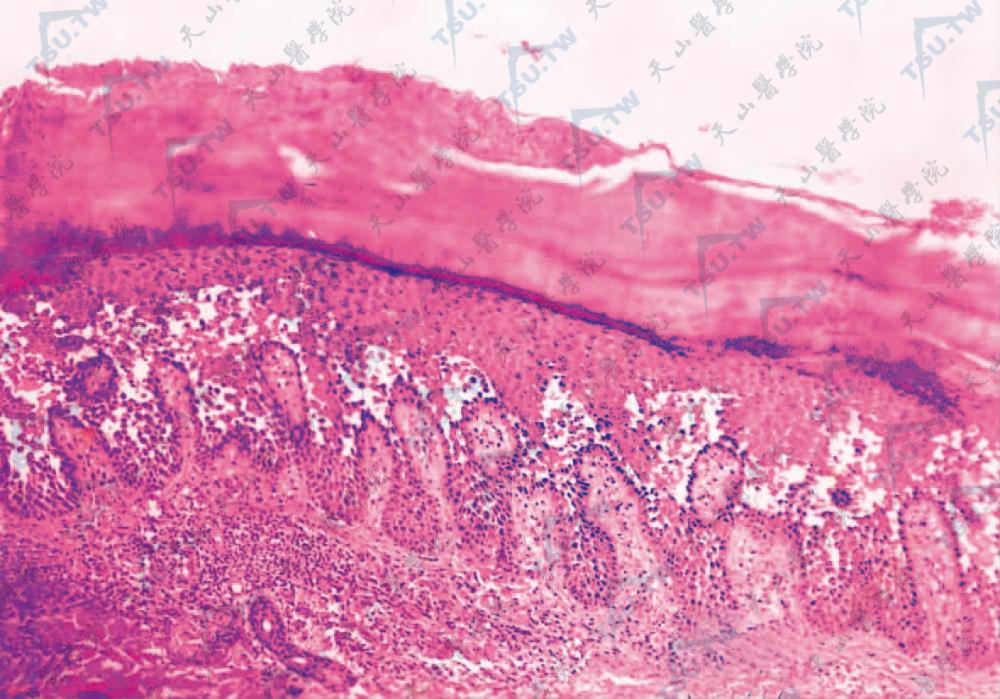
基底层上水疱,棘刺松解不完全,疱底绒毛增生(HE染色×100)
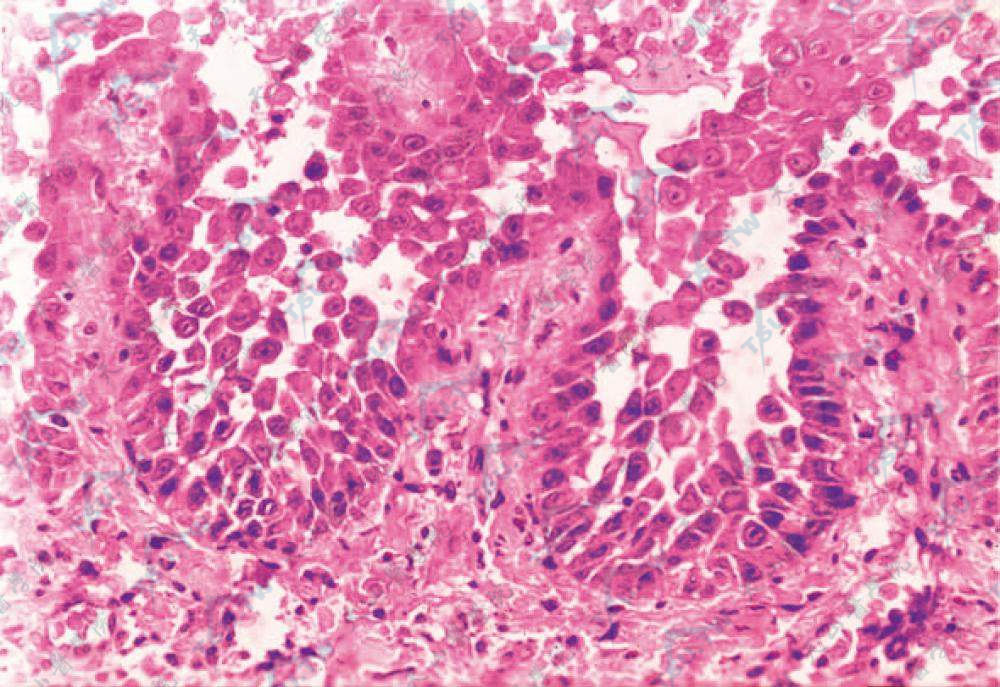
棘刺松解见单层基底细胞乳突(绒毛)向上突入裂隙内(HE染色×400)
诊断及鉴别
根据家族史,临床表现结合常规病理检查、免疫病理检查诊断不难。主要的鉴别诊断包括各型天疱疮、脓疱病和Darier病。
寻常型天疱疮和毛囊角化病虽然病理上与本病有相似之处,但临床上有差别。寻常型天疱疮口腔黏膜损害常见且严重。棘刺松解限于基底层上,棘刺松解细胞变性严重,不见角化不良细胞。直接免疫荧光棘细胞间有IgG沉积。
毛囊角化病皮损主要在脂溢部位发生角化性丘疹,常伴甲萎缩,病理为基层上方小的裂隙,不形成大疱,棘刺松解不显著,角化不良细胞明显。
复发性线状棘刺松解性皮病的临床和病理与本病相似,但其皮损限于身体一侧,且沿Blaschko线分布。好波及掌跖,皮损为红斑、水疱。家族性良性天疱疮一般不侵犯掌跖,其表现为点状小凹。
预防及治疗
本病病程较长,预后良好,50岁以后病情常减轻,但痊愈者少见。
治疗相对困难。系统使用有效的抗金黄色葡萄球菌的抗生素、外用抗生素药物或抗真菌药可得到改善。糖皮质激素外用、内服或两者合并使用也有效。严重者可用环孢素、口服维A酸和氨苯砜。皮肤磨削法和CO2激光气化治疗有效。特别严重者可进行皮肤移植。
