-
先天性耳瘘
本病又称先天性耳孔(congenital ear-pit),是指耳部于出生后即有一无症状的异常小孔或一小瘘管,常位于耳屏耳轮伸支上。病因及发病机制耳瘘或耳前瘘是由于胚胎发育过程中第1及第2
1 -
先天性下唇瘘
唇瘘、唇凹或先天性下唇凹是一种疾病,特征为在唇缘处下唇中央的两边呈双环状或裂缝样凹陷。瘘管中央深达5~25mm入唇内。通常伴有唇裂和腭裂和(或)耳前瘘道。本病由van der Woude
2 -
副耳
本病主要表现在耳屏前有小的皮色残片或结节,故又称耳赘(auricular appendages)、软骨性痣(nevus cartilaginus)或颈耳(cervical auricle)。副耳起源于耳结节或围绕第2、第3及第4腮
3 -
鼻横沟
本病又名鼻横线(stria nasi transuersa)。本病是一种少见的家族性缺陷,在鼻翼部发生横向的浅沟,主要由于鼻翼及鼻中隔软骨分化发育所致,可能为常染色体显性遗传。临床症状在婴儿
9 -
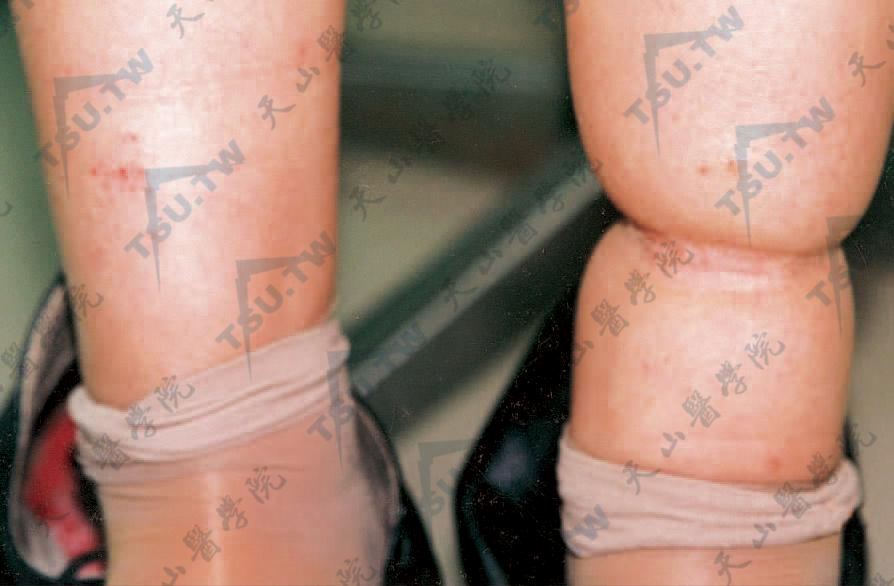
子宫内引起的缩窄带
本病是一种较少见的发育缺陷,主要是由于胎儿在宫体内时羊膜异常所致。有人亦称此病为羊膜带综合征(amniotic band syndrome)。本病临床表现主要是小腿、前臂或指部呈1~3mm宽、2~4
10 -
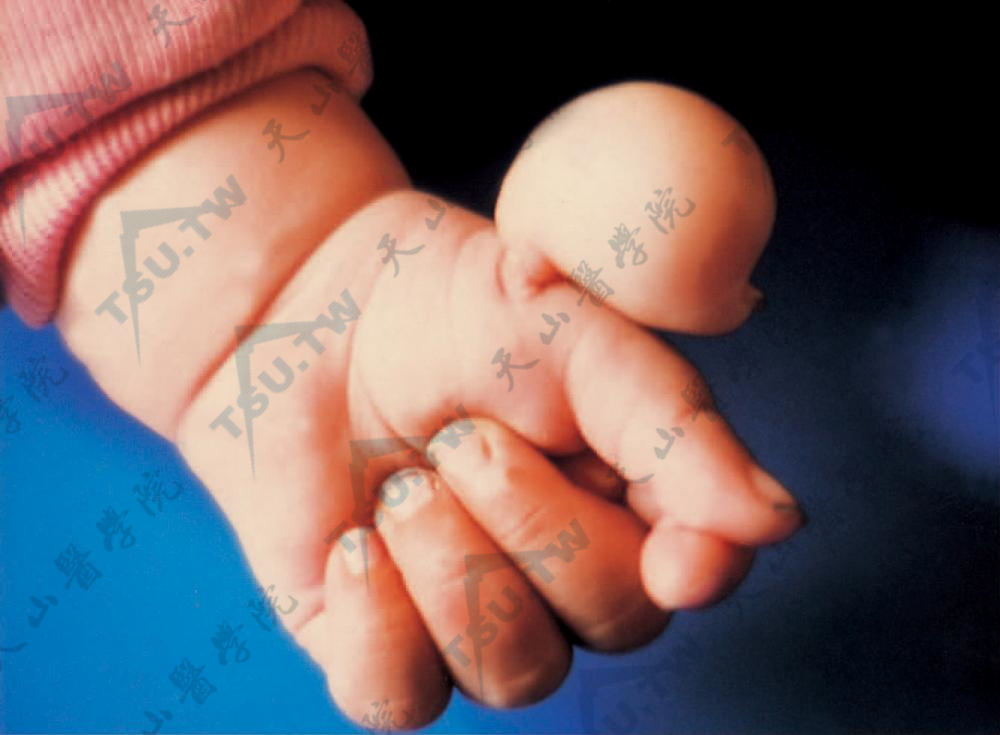
杵状指
本病又名Hippocratic指或肢端粗厚症(acropachy),是一种先天性或获得性指端畸形。病因及发病机制先天性者可能与不同外显率及基因表现的常染色体基因有关。很多获得性者遗传素质
11 -
遗传性并指症
本病常与手掌的Dupuytren挛缩症相混淆,偶或两病可同时出现,也有报道称本病是Marfan综合征及眼齿指增生不良症的一部分,与常染色体显性遗传和X连锁遗传均有关系。本病与手掌的Du
13 -
成人早老症
本病1940年由Werner首先报道,又名维纳综合征(Werner syndrome),是一种以代谢和结构异常为特征的早衰综合征,累及皮肤、毛发、眼、肌肉、脂肪组织、骨、血管和糖代谢。病因及发病
14 -
儿童早老症(哈钦森综合征)
儿童早老症(Progeria)又称哈钦森综合征(Hutchinson-Gilford syndrome)。Hutchinson于1886年首先报道。本病常发生在儿童,是以生长发育迟缓及在婴儿时期就发生进行性老年性变化为
15