
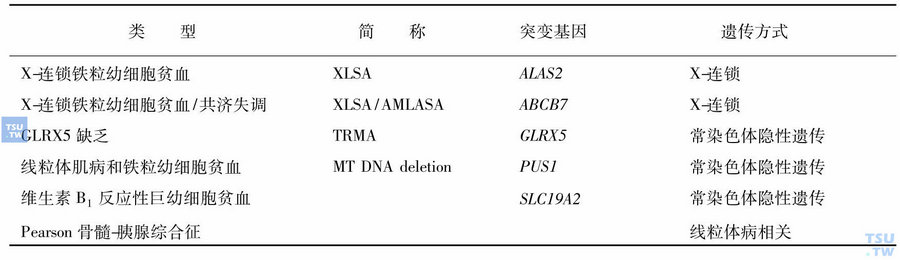
遗传方式
X染色体连锁伴性遗传(XLSA)
本类型最为多见,患者多为男性,女性携带者由于正常等位基因抑制了病态基因的表达,红细胞异常较少见。XLSA主要涉及两种基因突变:①δ-氨基γ酮戊酸合成酶2(ALAS2)基因突变所致的遗传性铁粒幼细胞贫血约占50%以上,限速酶ALAS 2仅表达红系细胞线粒体内,受细胞内铁水平调节,而不受血红素的反馈抑制。红系特异性ALAS2基因定位于Xp11. 2上,全长约22kb,由11个外显子构成,外显子5~11编码C末端高度保守部分,编码蛋白活性区约440个氨基酸,该区域出现突变会影响其蛋白的生物学活性。近年已陆续报道了超过25种XLSA的ALAS2基因突变类型。②X-连锁伴性遗传的XLAS与涉及小脑共济失调的ATP-结合亚家族ABCB7基因错义突变有关,该突变可导致铁硫簇生物合成的线粒体蛋白(包括ABCB7蛋白)功能缺陷。
常染色体遗传
常染色体遗传包括3种形式:①涉及GLRX5基因的GLRX5缺陷性SA;②涉及SLC19A2基因的维生素B1反应性、巨幼细胞性SA,SLC19A2基因编码维生素B1转运体,可导致胰岛素依赖型糖尿病、感觉神经性耳聋及巨幼细胞性SA,此病对维生素B1有部分治疗反应;③涉及假尿苷酸合酶1(PUS1)基因分子缺陷的线粒体肌病性铁粒幼细胞贫血(MLASA),PUS1缺陷可导致tRNAs改变,此缺陷普遍累及骨骼肌肉和血液系统。
线粒体病伴发遗传
Pearson骨髓-胰腺综合征为致死性疾病,因线粒体DNA缺失或重排所致的遗传性铁粒幼细胞贫血,多为散发或初发。贫血多为红细胞成熟障碍的正细胞或大细胞性贫血,带有空泡不成熟的幼红细胞和髓系细胞增多,以及大于50%的环状铁粒幼细胞,部分病例血小板及中性粒细胞减少;本病常伴有代谢性酸中毒、共济失调和胰腺外分泌障碍;较少幸存者进展为Kearns-Sayre综合征,表现为神经肌肉功能障碍、心脏传导系统异常、色素性视网膜病和铁粒幼细胞贫血,预后凶险。
遗传性铁粒幼细胞贫血的分类见表
发病机制
血红素合成
血红蛋白的合成需要铁进入线粒体合成血红素,而血红素的合成为一多步骤过程:首先甘氨酸及琥珀酸辅酶A聚集,在具有生物活性的辅酶5-磷酸吡哆醛(PLP)参与下,经ALA合成酶(ALAS)催化合成δ-氨基γ-戊酮酸(ALA),ALA转运至细胞质中合成血红素,然后血红素通过粪卟啉原Ⅲ转运回线粒体中,最后在亚铁原卟啉合成酶(FECH)催化下,铁插入原卟啉IX(PPIX)环中。机体可根据细胞内铁浓度,通过调控ALAS2、铁调节蛋白(IRP)、FTH1 和FTL的表达水平来调节铁吸收。ALAS2基因突变可导致细胞内铁吸收异常与血红素合成障碍。
铁硫簇合成
铁硫簇是多个代谢旁路激活的数个蛋白辅基,它对于线粒体功能尤为重要。铁硫簇生物合成和组装过程需要半胱氨酸脱氢酶(Nfs 1p)、伴侣蛋白(Isu1p/Isu2p)、Arh1p及Yah1p等氧化还原酶。铁硫簇主要通过降解IRP1、IRP2、转铁蛋白受体1 (TfR1)调节细胞内铁的水平。铁硫簇旁路某一蛋白失活可导致严重的线粒体内铁负荷过多,铁硫依赖酶活性缺失和氧化损伤,并最终进展为铁粒幼细胞贫血。铁硫簇基因Frataxin等突变可导致Friederich共济失调、神经变性、心肌病,部分患者出现色素缺乏性视网膜病。GLRX5是铁硫簇生物合成途径的一部分,其缺失导致细胞不能合成足够铁硫簇,胞质内顺乌头酸酶减少,增加IRP1对ALAS2和铁蛋白活性效应,致铁粒幼细胞贫血铁负荷过重。
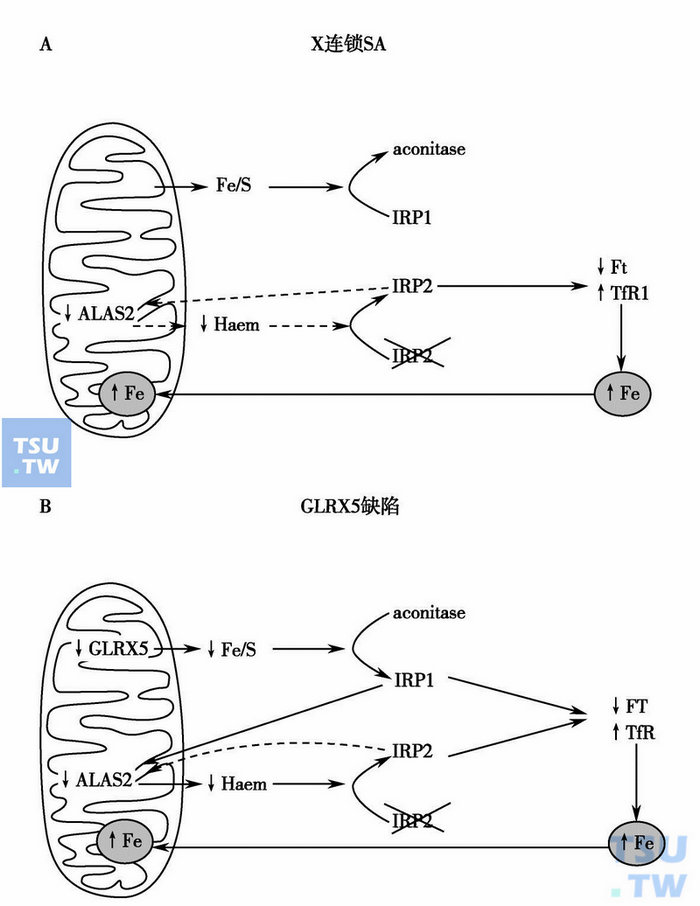
环形铁幼粒细胞贫血的发病机制
A. ALAS2基因缺陷减少血红素,IRP2增加铁饱和度,线粒体铁转运增加导致ALAS2进一步减少;B.红细胞内GLRX5缺乏引起IRP1增加和ALAS2转录抑制,血红素合成减少对IRP1的降解减低,红细胞内IRP1和IRP2联合作用增加转铁蛋白受体1(TfR1)的铁饱和度和线粒体铁转运。注:aconitase:顺乌头酸酶。
ABCB7异常表达
ATP结合盒转运子超家族成员ABCB7基因错义突变,ABCB7蛋白铁转运至胞质内功能受损,线粒体铁负荷过载,致X连锁铁粒幼细胞贫血伴小脑共济失调。
病理生理
正常骨髓中30%~50%的幼稚细胞经铁染色后,胞质中可见普鲁士蓝染色阳性的颗粒,这种细胞称为铁粒幼细胞,其胞质内颗粒为聚集的非结晶的三价铁磷酸盐和氢氧化铁,电镜观察发现此种颗粒既不在线粒体中,也不在胞质的其他细胞器中。当出现血红素合成障碍时,铁向细胞内转运并不减少,铁持续进入线粒体并蓄积其中。由于人类幼稚红细胞线粒体沿细胞核周围分布,铁染色后显微镜检查可见这种红细胞中普鲁士蓝染色阳性的铁小粒绕核呈环形分布,这种异常铁粒幼细胞称为环形铁粒幼细胞。电镜下可见粉尘状或斑块状含铁微粒在线粒体嵴间大量沉积,线粒体扭曲、肿胀,线粒体嵴难以辨认。
由于血红素合成障碍继发了血红蛋白合成障碍,红细胞呈低色素。体外实验中加入血红素后血红蛋白的合成得到纠正。细胞动力学研究表明本病出现贫血的主要因素为无效红细胞生成,表现为骨髓红系增生旺盛、网织红细胞正常或稍高,红细胞铁利用障碍,平均红细胞寿命正常或轻度缩短,轻度高胆红素血症及尿胆原排泄增加。
临床表现
一、乏力、苍白与小细胞低色素性贫血:XLSA多为男性,常染色体遗传家族女性亦可发病,杂合子及女性基因携带者一般无贫血,但红细胞形态多为小细胞性。大多数患者发病年龄在10~20岁,偶有50岁以上发病者。常见早期症状面色苍白、乏力、软弱,贫血多为中度。贫血轻微家族成员经家系调查后方可识别,不做家系调查可能漏诊。严重贫血可致幼儿及少儿生长发育迟缓。
二、铁负荷过载:慢性输血患者的严重并发症。无输血史但伴血色素基因(HFEC282Y)突变患者可发生更为严重的铁负荷过载。约1/3的患者出现糖尿病、皮肤色素沉着、肝功能轻度异常,以及一些不常见的感染如耶尔森肠炎菌所致的小肠结肠炎和毛霉菌病等。
三、共济失调相关的X-连锁铁粒幼细胞贫血(XLSA/A;OMIM301310):XLSA/A可于婴儿或儿童时期出现神经系统症状,表现为运动与认知能力障碍,主要临床表现为小脑共济失调、轻度小细胞低色性贫血和骨髓铁粒幼细胞增多,共济失调可发生于出生后第一年,以后不再进展;深肌腱反射亢进,巴宾斯基征可阳性。
四、骨髓-胰腺综合征:患者出生后不久即发病,伴胰腺外分泌功能不全,晚期发生肝肾衰竭。
实验室检查
贫血多为小细胞低色素性。网织红细胞正常或轻度增高,白细胞、血小板数多正常,红细胞渗透试验增强或降低。X-连锁遗传的铁粒幼细胞贫血的红细胞有二形性。常染色体隐性遗传铁粒幼细胞贫血的红细胞大小不均,严重者呈异形幼红细胞血症。线粒体病相关的铁粒幼细胞贫血多为大细胞贫血,外周血可见嗜碱性点彩红细胞、异形红细胞增多及有核红细胞。骨髓中红系细胞过度增生,可见双核原红细胞,可伴粒系、巨核系病态造血;铁染色显示铁粒幼细胞高达80%~90%,环形铁粒幼细胞大于15%。血清铁、转铁蛋白饱和度及铁蛋白均增高,肝活检显示铁质沉积与细小结节状肝硬化,提示储存铁增高。红细胞生存时间正常或稍缩短,未结合胆红素可增高。X-连锁铁粒幼细胞贫血红细胞原卟啉正常或减低,ABCA7红细胞原卟啉增高。X-连锁SA患者常合并血色病等位基因HFE(C282Y)突变。
诊断与鉴别诊断
根据本病特征:
- 小细胞低色素贫血;
- 红细胞明显大小不均;异形、靶形、椭圆形和点彩红细胞增多;
- 骨髓中红系细胞过度增生,铁染色显示含铁血黄素显著增多,铁粒幼细胞增至80%~90%,并可见到约10%~40%的环形铁粒幼细胞;
- 伴有无效红细胞生成;
- 血清铁大多增高,铁饱和度常显著增加,肝活检显示铁质沉积;
- 骨髓-胰腺综合征患者应伴有胰腺外分泌功能不全;
- 发病年龄轻等,铁粒幼细胞贫血不难诊断;临床病史特点有助于进一步鉴别特发性与继发性铁粒幼细胞贫血。另外部分地中海贫血患者因珠蛋白合成显著减少,多余的血红素可反馈抑制ALA合成酶,可同时伴发铁粒幼细胞贫血,应注意鉴别。遗传性AS需行必要的基因检查以鉴别其类型。
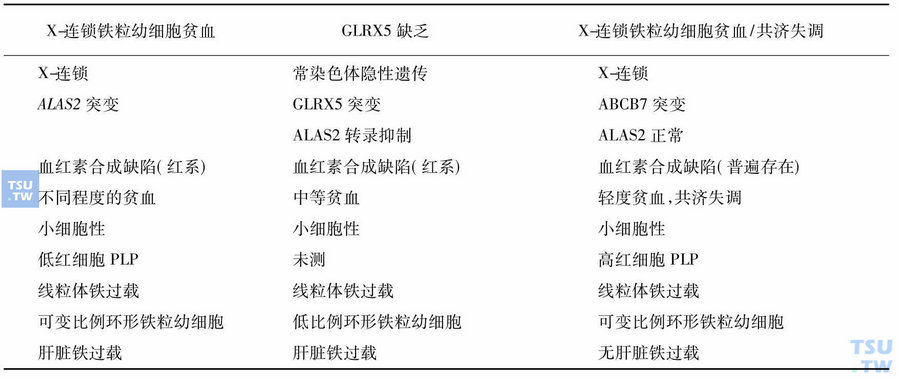
几种不同形式遗传性铁粒幼细胞贫血的鉴别
治疗
一、维生素B6凡诊断为本病者均应试用大剂量吡哆醇(维生素B6),100~200mg/d;约有不到半数的病例能减轻症状,XLSA可能对口服维生素B6更为有效。没有足够的证据表明胃肠外给药或用PLP-活性形式的辅酶比口服吡哆醇更有效。有效者网织红细胞增高,血红蛋白上升至正常或接近正常,或稳定在一个低于正常的水平,血清铁减少,FEP增高至正常,色氨酸代谢也得以纠正。然而红细胞的形态异常、低色素和环形铁粒幼细胞没有完全消失。有效病例的疗效不是一成不变的。有效者必须给予维持治疗,停药后几个月内即可复发。复发后再用吡哆醇治疗有时仍有效,但往往疗效不如第一次,有的则变为无效。加用左旋色氨酸有时可使吡哆醇治疗再有效。个别患者症状缓解后维持治疗一段时间可以停药。
二、去铁治疗:血红蛋白低于70g/L输血治疗依赖者,以及10次输血以上其血清铁蛋白大于1000g/L者应行去铁治疗,以避免糖尿病、肝硬化、性腺功能低下及心功能不全等并发症。维生素B6治疗后的XLAS可进行放血治疗。宜选择去铁胺皮下持续缓慢泵入或静脉缓慢泵入,剂量为20~40mg/kg,连续12~16小时泵入,每周5~7天。每周去铁治疗可增强维生素B6的疗效。GLRX5缺乏者行去铁治疗可提高其血红蛋白水平。
三、脾切除术:本病脾切除后极易发生血栓并发症且常为致命性的。除了由继发的血小板增多导致栓塞外,还有其他致栓因素,控制血小板数和其他抗凝治疗常无效,因此不宜行脾切除术。
四、骨髓移植:可根治本病,移植前应充分去铁治疗。
五、目前对骨髓-胰腺综合征尚缺乏有效治疗手段。
预后
对吡哆醇治疗有效患者能较好的生存多年,无效者多因骨髓衰减、严重贫血、心律失常、肝功能衰减或继发感染而死亡。骨髓-胰腺综合征缺乏有效治疗方法,预后较差。
