
G蛋白是鸟核苷酸结合蛋白(guanine nucleotide-binding protein)的简称。根据结构和功能,G蛋白分为3类,即异三聚体G蛋白(Gs)、小分子量单体G蛋白和大分子量G蛋白。与内分泌代谢性疾病关系密切的主要是异三聚体G蛋白,它是G蛋白耦联受体的关键信号转导物,缺乏G蛋白者不能存活。异三聚体G蛋白分子含内源性GTP酶活性,能水解GTP生成GDP,并通过GTP/GDP转化进行G蛋白循环。
G蛋白α亚基差异是保持信号转导特异性的基础
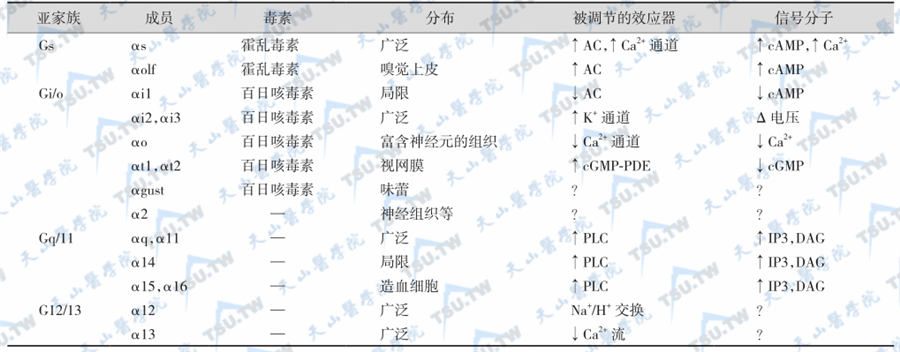
人们习惯地将Gα亚基分为4个亚族。由于编码Gα产物的剪接方式不同,可生成多种不同的亚型Gα。G蛋白由α、β和γ等3种亚基组成,总分子量约100kD,其中与GTP/ GDP结合的Gα亚基的分子量为39~45kD。G蛋白超家族的成员很多,因其α亚基能与GTP结合,各种G蛋白的主要差异在α亚基。目前已确定结构的Gα有20多种,Gβ有5种,Gγ有12种。因此,机体组织中的G蛋白异三聚体的组合方式可超过1000种以上,这是保持G蛋白信号转导特异性的重要原因。
Gα亚基的分类
同类型的GPCR-G蛋白下游信号转导途径有所不同,主要有数条共同通路,如腺苷环化酶-cAMP-蛋白激酶A通路、G蛋白-MAPK通路、磷脂酰肌醇-Ca2+信号通路、DAG-蛋白激酶C通路等。因而G蛋白下游的信号转导有4个显著特点:
- 相对GPCR和GPCR配体的数目来说,G蛋白下游的信号通路要少得多,因而,激素-受体的特异性是相对的,有时由于效应与信号转导分子相互作用,可出现激素特异性丢失现象;
- GPCR和生长因子、细胞因子之间可通过各自和共同的信号转导系统相互“串语”,其中最明显的结果是引起ERK/MAPK磷酸化级联反应的激活和生长因子受体的反式激活;
- GPCR被激活后需要先形成同(异)二聚体才能使信号被G蛋白“接收”(G蛋白激活),但也存在一些例外情况。
目前已能用GPCR的特异性cDNAs、抗体技术及一些生物物理技术(如生物发光或生物荧光法与能量转移法等)揭示GPCR的激活机制。GPCR被激活后的多价螯合(sequestration)是受体被再次“致敏”的前提,同时也是受体“内陷”的基本步骤,而GPCR激酶(G protein-coupled receptor kinase,GRKs)和SS-arrestin则为GPCR的“脱敏”因子。而这些过程都受辅因子的调节。因此,上述的经典G蛋白下游途径只是信号转导体系中的共有通路,它们可能是信号转导的“主干”,而在主干之外,这些辅因子则组成“旁支”,形成一个复杂的信号调节网络;许多药物能直接或间接作用于GPCR或其下游信号分子,因此GPCR药物必须用生理变化指标来评价药物的有效性。
G蛋白复合物转导G蛋白耦联受体信号
异三聚体G蛋白(Gs)能耦联GPCR与其效应体——腺苷环化酶。G蛋白的α亚基(Gsα)突变(如R321H、R265H、S250R等)导致Gs耦联信号转导途径活化,体细胞Gs的这类活化性突变常常是一些内分泌肿瘤、骨纤维性增生不良症和McCune-Albright综合征的病因;相反,Gsα(又称为GNAS1)杂合性失活性突变常导致Albright遗传性骨营养不良症(Albright hereditary osteodystrophy,AHO)和进行性骨异常增生症(progressive osseous heteroplasia)。由父方遗传的GNAS1突变仅表现为假-假性甲旁减症(pseudopseudohypoparathyroidism 1α,PPHP),没有对PTH不敏感的表现;而由母方遗传时,还加有其他激素(如TSH、PTH)不敏感的表现。具有PPHP表型的AHO母亲,其杂合型错义突变遗传给其女儿时,女儿却表现出PHP1α的表型。在Gsα基因剔除小鼠的研究中发现,两个等位基因在多数组织中表达,但在一些组织(如肾远曲小管)只有母方来源的等位基因表达。母方来源的等位基因被毁使肾小管细胞不能表达Gsα,而父方来源的等位基因被毁对肾小管上皮细胞的PTH功能无明显影响。
G蛋白调节效应体功能
GPCR与激素(配体)结合后,激素-受体复合物使兴奋性G蛋白(Gs)或抑制性G蛋白(Gi)中的α亚基与三磷酸鸟苷(GTP)结合到复合物上,后者促进(或抑制)腺苷环化酶转变为cAMP,cAMP再与cAMP依赖性调节蛋白激酶的调节亚基结合,蛋白激酶被激活,蛋白磷酸化并改变细胞的功能。另一方面,激活的催化性蛋白激酶进入细胞核,使与cAMP反应元件结合的蛋白磷酸化(激活)。完成信号转导后,磷酸二酯酶降解cAMP为5′-AMP。由此可见,G蛋白的功能是通过其α亚基与GDP或GTP的结合而完成对效应体的调节,随着GPCR的激活与失活所进行的循环性变化称为G蛋白循环。
G蛋白耦联受体分为A/B/C三个亚族
GPCR为7次穿膜受体,故亦称为7TM受体(seventransmembrane segment receptor),在第5及第6跨膜α螺旋结构之间的细胞内环部分,是与G蛋白耦联的区域。据估计,人体内的GPCR种类超过2000种,现已克隆出100多种GPCR,其配体主要为蛋白质,但也可以是胺类、多肽、糖蛋白、脂质、核苷酸、离子或蛋白酶。此外,外源性刺激物如光、气味也可以GPCR为介导,将信息转给靶细胞。
各种G蛋白耦联受体激活途径不同
GPCR分子相互作用
许多GPCR含有自动激活活性,在缺乏配体情况下,可将GPCR激活。用人工方法将肾上腺素能受体第3胞内环袢的C端部分替代可引起体质性(constitutive)受体活化;之后发现,自然和人工发生的活化性GPCR突变部位并不只是局限于此区,其他很多位点的突变也可引起受体自动激活。Kjelsberg等将α1b肾上腺素能受体胞内第3环袢Ala293残基用所有其他可替代的氨基酸替代,均发生体质性活化。这一实验的意义在于,它间接证明了一个事实,即在GPCR分子中存在1个或多个限制受体分子自动激活的结构域,一旦其功能丧失,即可改变分子激活的自限能力,导致受体自动激活,这一(些)限制位点一般是分子的关键氨基酸残基或片段。它的作用是阻滞分子内部各片段相互作用,只有当配体与受体结合时,才可解除对分子激活的阻滞作用。同理,GPCR的活化性突变分子机制之一是突变型受体丧失这种能力,使受体分子在缺乏配体情况下,处于激活状态(分子的某些关键部位与G蛋白接触,启动下游的信号转导通路)。或者,突变型β2肾上腺素能受体的空间构象不稳定,使活化型与非活化型受体构象的转换比野生型受体容易得多。例如,LH受体和FSH受体的第5与第6穿膜段的相互作用是维持受体非活化状态的关键因素,如果这些区域发生突变则导致受体自动激活。
GPCR分子质子化(protonation)
在GPCR的A亚族成员中,第3个胞内环袢中高度保守的Glu/Asp-Arg-Tyr(D/ ERY)基序在受体活化中起着重要作用(D/ERY基序中的门冬氨酸质子化)。例如,视紫红蛋白受体Glu134摄取质子时伴有光效应物生成和pH变化,转变为金属型视紫红蛋白受体Ⅱ状态。α1b和β2肾上腺素能受体突变具有电荷中和特点,门冬氨酸或谷氨酸的电荷被中和后,受体即转变为活化状态。GnRH受体的门冬氨酸残基突变也使质子化反应加强。
GPCR变构效应
GPCR中的组氨酸与Zn2+形成的交链对(cross-linking pairs)可抑制信号转导活性。这种现象见于视紫红受体、β2肾上腺素能受体和PTH受体。有些激素受体拮抗剂具有负性抑制活性,在一定条件下起到激动剂作用,这种激素受体配体称为反转性激动剂(inverse agonists)。激动剂和反转性激动剂与GPCR结合后,受体分子出现的变构效应是受体分子激活的关键反应。有些突变使受体分子处于变构效应状态,出现GPCR自动激活,或者在配体作用下,较野生型受体更易进入活化状态。参与变构效应的氨基酸残基很多,其中二硫键的作用十分重要,变构的关键部位一般发生在第3和第6穿膜段。A亚族中的各种受体变构过程十分相似。
GPCR信号向G蛋白的转导
GPCR被激活后,如何将信号转导给G蛋白的机制仍未阐明。一些研究表明,第2、第3胞内环袢以及C端的近段氨基酸残基是信号转导(即与G蛋白耦联)的结构基础,其中第3胞内环袢主要决定耦联的特异性而第2环袢主要决定耦联效应的强弱。在M5毒蕈碱受体中,胞内第2环袢α-螺旋的扩展肽与受体激活有关,而对侧残基与G蛋白耦联(激活G蛋白)有关。此外,GPCR信号向G蛋白转导还与DRY基序、受体胞内第2环袢的α-螺旋及环袢中的微结构域(microdomain)有关。
受体二聚化
现已证明,含酪氨酸激酶受体(receptor tyrosine kinase)的二聚化是该类受体信号转导所必需的。GPCR也形成同二聚体(如β2肾上腺素能受体,δ-阿片样受体、细胞因子受体、钙受体及谷氨酸受体等)。二聚体有利于维持受体密度和调节受体内陷过程,有些受体激活以及突变型受体出现的优势负性作用必须以受体的同二聚化为前提。另一方面,同一受体不同亚型(如GABAⅠ型受体)也必须先形成异二聚体能发挥功能。
G蛋白病涉及的范围广泛
生物通过进化获得多样性。G蛋白耦联受体与G蛋白参与激素作用、循环调节、神经传递、机体防御等多种信息网络功能。G蛋白在具备多样性的同时,还通过进化保存了相同或相类似的作用机制。G蛋白3个亚基基因突变所致的疾病称为G蛋白病(G protein diseases),G蛋白病的疾病谱广,病种多,已从内分泌疾病扩展至其他专科,而且还在不断发现新的病种。目前发现的主要是G蛋白α亚基基因突变,而对G蛋白的β亚基与γ亚基尚缺乏深入研究。但是,β与γ亚基在信号转导、细胞反应、靶基因转录和细胞凋亡的调节中也同样起着重要作用。例如,β亚基含有色氨酸-门冬氨酸为尾端的WD重复序列(通常为4次或更多次重复)的突变或变异与一些常见疾病(如高血压、冠心病、胃肠疾病、呼吸道疾病、神经精神疾病等)有着密切的病因关系。同时,G蛋白与药物的作用机制及G蛋白的相关药物的遗传差异又是遗传药理学研究和新药物开发的重要课题。
另一方面,G蛋白耦联受体和类固醇激素的核受体又有密切联系,该类激素均有相应的膜受体。其中部分膜受体即属于G蛋白耦联受体。例如,雌激素是一种经典的类固醇类激素,但近年发现,雌激素的膜受体GPR30即为G蛋白耦联受体家族成员。
Gα基因活化性突变
一般导致G蛋白功能亢进。从化学反应上看,G蛋白失活是通过Gα的GTP酶水解GTP 成GDP所致;而从基因上看,GTP酶反应的Gα基因突变(主要是精氨酸和谷氨酰胺的替代突变)可抑制GTP酶活性,使Gα呈维持开启状态,出现G蛋白功能亢进。
GH瘤和Albright综合征是G蛋白功能亢进的典型例子。G蛋白信号调控因子(RGS)的作用是使GTP结合Gα,进而使GTP酶活性增强10~100倍。目前发现至少16种RGS,它们选择性作用于Gi、Gt、Gq、G13,使Gα关闭在10秒至1秒以下。在迅速调控心肌收缩(Gi)、视觉(Gt)、血管收缩(Gq)同时,对调控激动剂的敏感性也有重要作用。今后有可能在Gs激动剂敏感性疾病(如心律不齐、高血压等)中发现更多类型。
G蛋白失活性突变
主要引起G蛋白功能减退。这类疾病主要有假性甲旁减(PHP Ⅰa、PHP Ⅰb和PHPⅠc)和假-假性甲旁减(PPHP)。例如,1个Gsα等位基因功能丧失可引起PHP Ⅰa。Gsα基因位于染色体20q13,从PHP Ia家系中已发现16个以上的Gsα基因异常。PHPⅠa为常染色体显性遗传性疾病,其病因是:①对甲状旁腺激素(PTH)抵抗;②Gs介导的多数激素抵抗;③Albright遗传性骨营养不良症(AHO)所组成的各种综合征。
Gsα基因具有特异性接纳父传基因的特征,这是Gsα功能丧失引起PHP/PPHP的原因。人的PHP/PPHP家系遗传学分析证明,母传的是PHP而父源的是PPHP。Gsα基因剔除的结果与此相符,即剔除Gα基因的纯合子动物不能存活,而杂合子则导致PTH抵抗。也就是说,Gsα的1个等位基因突变如来源于母亲可发病,来源于父亲却不发病。父传Gsα基因表达可因基因印记而抑制。因此,Gs异常在母传时的活性为零,产生PHP;父传时,Gs活性正常而成为PPHP。此外,在与PHP、PPHP症状相同的AHO中,无Gsα基因印记,Gs活化降低50%,但无临床表现。
PHPⅠb患者对PTH不敏感,但不伴AHO。从PHPⅠb家系的遗传学分析结果显示,致病基因由父传的等位基因印记调控,定位于Gsα附近的20q13.3,推测PHPⅠb的原因在于Gsα上游缺陷。
G蛋白的种类和功能差异
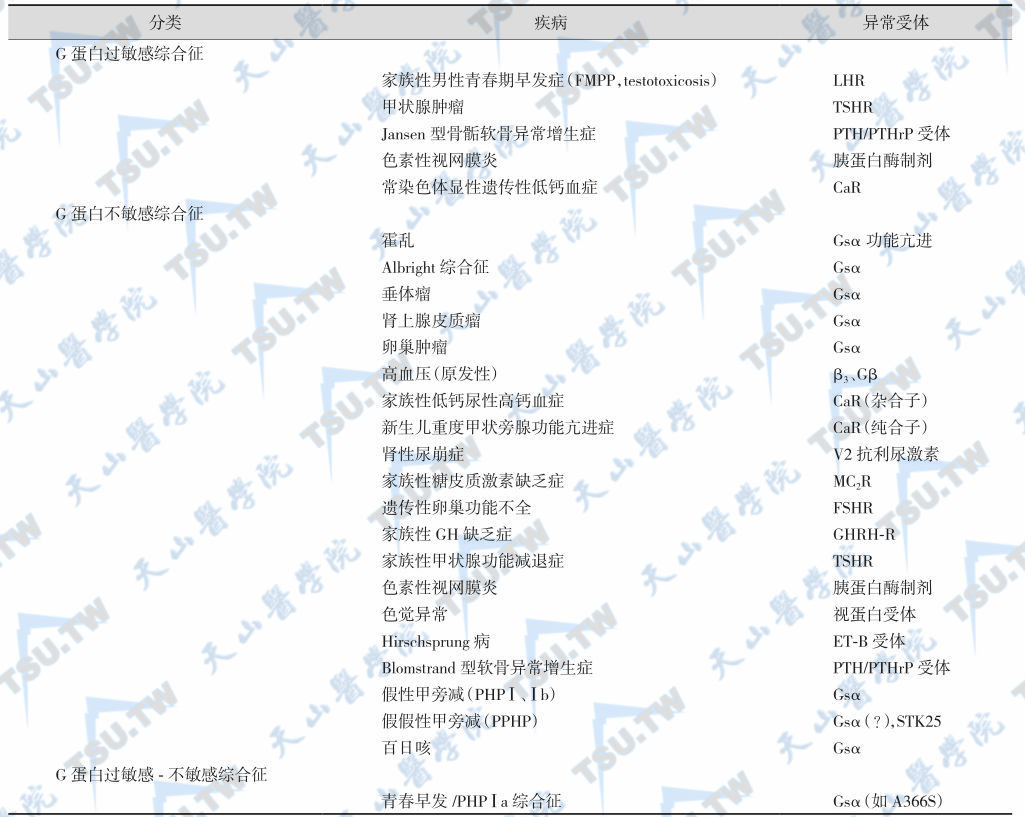
常导致G蛋白功能亢进-功能减退综合征,见于垂体选择性甲状腺激素不敏感综合征和假性甲旁减(Ⅰa型)伴性早熟或青春期早发等。G蛋白功能异常所致的常见疾病见下表。
激素敏感综合征和激素不敏感综合征
多基因疾病与G蛋白功能异常
一、原发性高血压
各种循环调节激素引起的平滑肌收缩均由钙浓度(钙敏感性)调节,而钙敏感性调节中的低分子量G蛋白Rho及Rho激酶(p160ROCK)发挥着重要作用。抑制Rho的肉毒C3毒素和p160ROCK的拮抗药可抑制钙依赖性肌肉收缩。即Rho经p160ROCK介导,作用于肌凝蛋白,调节钙敏感性。此外,p160ROCK在平滑肌以外的细胞也有活化Na+/H+交换体(NHE)作用。p160ROCK拮抗药使大鼠的高血压得到控制,因此推测Rho与生理性血压调节及高血压病的发病有关。
GNB3基因编码G蛋白的β3亚类,该基因825位C→T突变使mRNA剪接位点移动,G蛋白信号转导功能增强。该突变与高血压、肥胖和左心室肥厚有关,而且,在轻度高血压病人中,GNB3基因825T突变与心室肥厚有直接关系。
二、 钙受体病
CaR胞质内结构域990位点多态性(A/A、A/G、G/G)与第4号内含子的T→C突变(T/T、T/C、V/T)和PTH分泌及血清钙有一定关系。在血液透析病人中,A/A组的血清PTH明显高于G/G组和A/G组。因此,分析透析病人CaR多态性有助于预测继发性甲旁亢的严重性。CaR基因(位于3q)的杂合子失活性突变引起家族性甲旁亢(FPT)和家族性良性低尿钙性高钙血症(FHH)。这两种疾病可能与CaR基因点突变(如F881L,位于受体的胞质部分的尾部)有关。突变型CaR在人胚肾细胞中表达,使Ca2+的调定点升高,引起钙的重吸收增多和高钙血症。FHH为一种良性综合征,尿钙排出降低,血钙升高,血清PTH正常或升高(不被抑制)。
CaR基因活化性突变导致低血PTH性低钙血症。病人在治疗前的低钙血症较轻,而治疗后尿钙排出明显增多,其反应性明显高于特发性甲旁减病人。如低钙血症病人的尿钙/尿肌酐比值相对正常,要高度怀疑CaR基因活化性突变可能,这也是与特发性甲旁减鉴别的重要临床依据之一。CaR功能缺失突变的杂合子导致家族性低尿钙性高钙血症(familial hypocalciuric hypercalcemia,FHH),而纯合子则表现为新生儿严重甲状旁腺功能亢进症(NSHPT);相反,CaR功能获得性突变则可以导致家族性高尿钙低钙血症(常染色体显性散发性甲旁低)。这种CaR功能获得性突变一般都发生在受体的胞外部分(N端)。FHH高钙血症的原因是CaR对钙离子调节PTH的调定点升高,NSHPT则是调定点升得更高,使甲状旁腺对Ca2+表现出“抵抗”所致。高钙时CaR活化某种关键信息传递通路,进而抑制PTH分泌的机制仍不明了。
三、多囊肾
常染色体显性遗传性多囊肾是由于PKD1 和PKD2基因突变所致,多囊素-1(polycystin-1)和-2为PKD1和PKD2的表达产物,PKD受体为GPCR成员。患者的肾小管扩张,严重时形成囊肿,囊肿中的ATP含量升高。细胞中的PZY-GPCR和PZX嘌呤受体(purinergic receptor)表达异常可能是囊肿扩张的促发因素。
四、肥胖
基因组印迹(genomic imprinting)决定遗传表象(epigenetic phenomenon),使父母的两个等位基因产生不同的表达,而印迹基因(imprinted genes)调节胎儿的生长发育和出生后的代谢,现证明,Gsα是决定个体的代谢类型和特性的主要印迹基因之一。如果母方的Gsα异常可导致肥胖和其他相关性疾病。
(周后德)
