
PaCO2升高的潜在不良作用见表2-24。比较重要的临床问题大多数发生在PaCO2水平高于150mmHg时。然而,PaCO2即使少量的增加,也增加脑血流,因此当颅内压增高(例如急性头颅损伤)时,“允许高碳酸血症”策略一般是禁忌的。PaCO2的增高也刺激通气,但在实施“允许高碳酸血症”策略时,患者通常已应用镇静剂,故刺激通气的作用在临床上常不明显。

允许高碳酸血症对某些患者的氧合有不良影响。PaCO2增高和pH降低使氧合解离曲线右移,这降低了血红蛋白对氧的亲和力,也就减少了肺内血流的携氧量,但有利于氧在血红蛋白中解离,便于组织摄取。根据肺泡气体等式可以说明,肺泡PCO2的增高可导致肺泡PO2的降低,PaCO2每增高ImmHg,PaO2就降低约1mmHg。实施允许高碳酸血症时,理想的话,应努力使氧合达到最大。
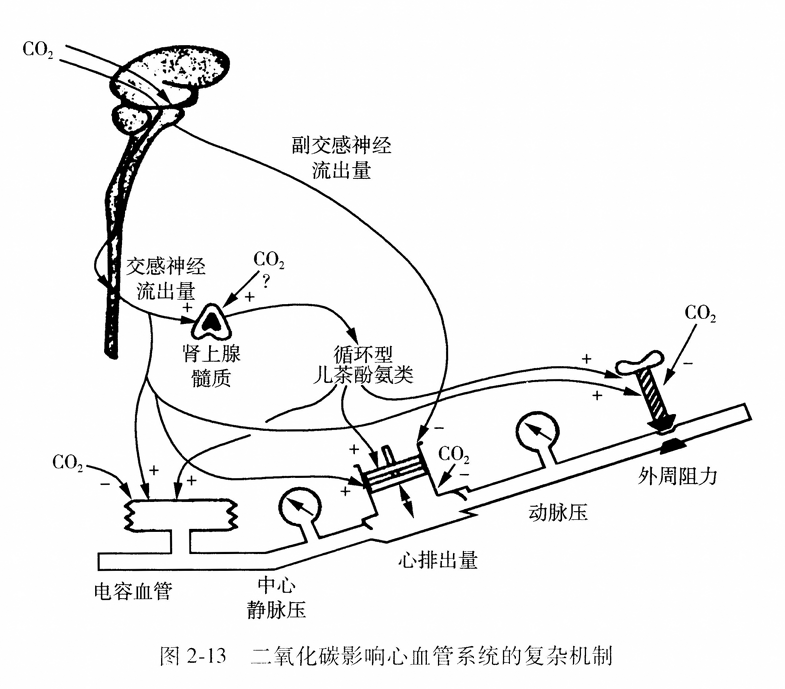
二氧化碳对心血管系统的影响难以预测,如图2-13所说明的,二氧化碳诱发对心血管系统的互相对抗性反应,二氧化碳直接刺激或抑制心血管系统的某些部分,经过对自主神经系统的刺激而发生相反的作用,因此难以准确预计心血管系统对允许性高碳酸血症的反应。我们的经验,PaCO2的增高最常引起肺动脉高压,心排出量增加,作用于心血管和自主神经系统的药物剂量在允许高碳酸血症时需要调整,但这是酸中毒而不是PaCO2升高的结果。
限制允许性高碳酸血症的主要因素是pH的改变。没有原发心血管疾病或肾衰竭的患者常能耐受7.20~7.25的pH,年轻患者甚至可耐受更低的pH,但究竟能耐受多低的pH则依每位患者的基础状况而定。应努力设法让PaCO2从机械通气开始实施允许高碳酸血症策略时起逐渐增加,而不是突然迅速增加,以便让肾能逐渐代偿。通气策略(呼吸机参数)的突然大幅改变,会导致PaCO2的明显增高和pH的迅速降低,增加患者的不耐受性。
在处理允许高碳酸血症引起的酸中毒时,对是否应用碱性药物尚有争论。在心脏骤停时,碳酸氢钠是禁用的,因为这可导致细胞内酸中毒的加重。然而在允许高碳酸血症时能否应用,则还没有广泛的研究。当给予碳酸氢钠时,我们希望的是,二氧化碳负荷只是短时间的增加,如果通气水平保持不变,在经过一段时间后,二氧化碳就会被呼出。但是,应用碱性缓冲剂,对允许高碳酸血症整个耐受性是否有任何影响尚不清楚。有人主张以THAM代替碳酸氢钠,因为THAM不产生CO2,而对细胞内和细胞外的酸中毒有缓冲作用。
只有当气道压已达高限阈值(平台压<30cmH2O),通气频率已达到最大(如f达30~35次/分,直至产生内源性PEEP)时才推荐应用允许高碳酸血症策略。在大多数患者,实施允许性高碳酸血症对中枢神经系统在短期内似乎没有明显的不良反应,但是否有长期的不良反应则不清楚。
PHC的明显好处和良好耐受性已引起了对高碳酸血症后果的重新评价。高碳酸血症的重要作用是由细胞内pH调节的。现已明确,细胞内pH的改变,与急性高碳酸血症后细胞外pH改变比较,有明显不同的时间过程。CO2弥散自由通过细胞外和细胞内腔,在这两个腔内,急性高碳酸血症引起的PaCO2和pH改变在初始时是类似的,然而,细胞内pH在3小时内恢复正常的90%,而肾对细胞外pH的代偿性纠正缓慢发生,在3天后仍保持不完全代偿。这种细胞内pH的快速纠正是由于细胞内缓冲、有机酸消耗和细胞壁质子泵作用的结果。这有两个重要的影响:首先,急性高碳酸血症性酸中毒的许多作用,是由细胞内pH介导的,一旦发生代偿,即不再继续存在;其次,细胞外pH并不准确反映细胞内pH,因此它不是急性高碳酸血症性酸中毒作用的可靠指标。
急性高碳酸血症对心血管系统也有重要影响,表现为对心肌收缩力的直接抑制作用,以增加肾上腺素和去甲肾上腺素的释放来增加交感神经的兴奋性和直接扩张周围血管。多数有关对正常的或受损伤肺的高碳酸血症的研究显示氧合改善,分流系数减少,肺血管阻力增加,这些作用由酸血症所介导。
还有研究提示,相当严重的酸血症在血氧正常情况下引起很小的细胞损伤和对缺氧性损伤有保护作用,后一现象的机制可能是酸血症时细胞对氧的需要减少。
综上所述,虽然认为没有控制的高碳酸血症是有害的,但机械通气期间的控制性高碳酸血症或PHC似乎是比预想的能更好耐受,急性高碳酸血症如果没有达到非常高的PaCO2水平(>150mmHg),那么是和迅速的细胞内代偿,对中枢神经系统没有严重的不良反应相关的,在大多数情况下,对循环也没有严重的不良反应,并能改善动脉血氧合和可能减轻缺氧的细胞损伤。只要高碳酸血症的发生是比较缓慢的,有时间让细胞内pH发生代偿和应用适量的镇静剂,那么患者对高碳酸血症的耐受性似乎也是好的。
