
调节肽主要包括存在于神经系统的神经递质或调节物和存在于内分泌细胞起着循环激素或旁分泌激素作用的两类活性多肽。这些活性肽多由4~40个氨基酸残基组成,广泛分布于体内各器官组织。在生理条件下,调节肽对组织器官的运动、分泌、感觉、营养、代谢和防御等功能活动具有保护和调节作用,是维持机体内环境恒定的最重要的机制之一。
现已在心血管系统中发现了30多种调节肽。在这些调节肽中,有的是心血管系统肽能神经纤维分泌的神经递质,如降钙素基因相关肽(CGRP)、神a经肽Y(NPY)、速激肽(tachykinin,TKN)、血管活性肠肽(VIP)、神经降压素(neurotensin,NT)、阿片肽(opioid peptides);有的是心血管系统自身产生和分泌的一些激素,如ANP、肾素、血管紧张素和ET;有的则来自其他内分泌组织所产生的一些肽类循环激素,如TRH、CRH、ACTH、AVP、胰岛素、胰高血糖素、生长抑素、蛙皮素和缓激肽(BK)等。还有一些是从心血管系统提取出来的活性多肽,但其确切的定位和来源尚不清楚,暂命名为心血管系统的内调素(endocoids),主要有抗心律失常肽(antiarrhythmic peptide,AAP)、心脏兴奋肽(cardioexcitatory peptide)、心脏加速肽(cardioacceleratory peptide)、心脏活性肽(cardioactive peptide)和心肌生长因子(cardiac growth factor)等。这些调节肽不仅对心血管系统功能和生长发育具有重要的调节作用,而且对心血管系统疾病的发生、发展亦具有重要影响。
抗心律失常肽对抗心律失常
目前已可人工合成抗心律失常肽(antiarrhythmic peptide,AAP),并建立了放射免疫测定法。除心脏外,许多其他组织中亦含有AAP,但以心房组织中含量最高。心房的AAP含量随年龄而异,年龄越小,含量越低。心脏内的AAP有两种分子形式,大分子形式可能是小分子AAP的前体。血清中只有小分子形式的AAP。AAP可减慢心率,延长Q-T间期,对抗低钾、高钙和G毒毛旋花苷所引起的心律失常,具有强大的抗心律失常作用,优于维拉帕米,可能通过抑制Ca2+进入细胞和K+流出细胞而起作用。
AAP较稳定,静脉或腹腔注射均有效,口服也可被吸收。体内半衰期约10分钟,主要从尿中排出。静脉注射14CAAP,可迅速分布于体内许多器官,但不能通过血-脑脊液屏障,故不能进入脑和脊髓。许多抗心律失常药物如奎尼丁、利多卡因、普鲁卡因胺和维拉帕米等,其有效剂量与中毒剂量十分接近,且有一定的体内积蓄作用,使临床应用受限。而AAP的毒性极低,即使静脉注射有效剂量的40~1000倍,亦不影响血压和呼吸,也未发现毒性反应。此外,AAP还抗血小板聚集,有抗血栓作用,故可防止血栓形成。AAP对纤溶系统的活性、复钙时间和动脉血流量无明显作用。
降钙素基因相关肽与高血压相关
降钙素基因相关肽(CGRP)由37个氨基酸残基组成。体内CGRP与降钙素来自同一个基因,但其基因剪接和转录方式不同,故CGRP和降钙素是同一基因不同转录和表达的两种生物活性多肽。CGRP基因定位在第11号染色体的短臂上,由2800 bp组成,含有5个内含子和6个外显子。它们在不同的组织中进行基因重组,在甲状旁腺可以转录,表达成降钙素;而在神经系统可以转录,表达成CGRP。降钙素和CGRP虽然来自同一基因,但其结构、分布和功能各不相同。人的CGRP有α和β两种分子形式,其氨基酸的组成和序列基本相同,仅在第3、22和25位有区别(α为天门冬氨酸、缬氨酸和天门冬酰胺;β为天门冬酰胺、蛋氨酸和丝氨酸)。
CGRP在体内主要分布于神经系统,在心血管系统和肺组织内亦有广泛分布。在心脏,心房含量最高。CGRP的特异性受体主要存在于心房、心室和血管系统,其中以心房的密度最高。CGRP与受体结合后,激活腺苷环化酶,使细胞内cAMP水平升高发挥其生物学效应。在正常情况下,CGRP很难通过血-脑脊液屏障。有人发现若用脂质体包被CGRP,则可通过血-脑脊液屏障,透入脑实质。CGRP具有强大的舒张冠状动脉作用,其作用比硝酸甘油、硝普钠约强240倍,也远强于P物质、ANP和异丙基肾上腺素。CGRP舒张冠状动脉和脑血管的作用在去除内皮细胞时其舒张作用依然存在,不依赖内皮细胞的完整性。CGRP对粥样硬化的冠状动脉仍有舒张作用。CGRP舒张血管,降低血压的作用可能是通过对血管的直接抑制作用而起的,其作用不受预先使用肾上腺素受体阻断剂、胆碱能受体阻滞剂和组胺受体阻断剂、利舍平、去迷走神经或前列腺素合成抑制剂的影响;CGRP对心脏具有正性变力和变时作用,可使心率加快、心肌收缩力增强和心排血量增加。对心房肌的作用尤其明显,强于去甲肾上腺素;CGRP促进缺血心肌功能的恢复,抑制脂质过氧化,防止蛋白质和酶的漏出。此外,体内几乎所有血管外膜或平滑肌层均有CGRP的神经纤维分布,它是调节心血管活动的重要肽能神经纤维。此外,CGRP还可促进前列环素的释放和细胞内、外的Na+/Ca2+交换。
给原发性高血压和高血压脑梗死的患者应用CGRP,可有效降低血压,增加心排血量和脑及其他重要器官的血流量。这是因为CGRP具有拮抗ET收缩血管、升高血压等生物效应的作用,被认为是一种心血管保护因子。探讨CGRP的临床药理,延长CGRP体内作用时间和克服机体的快速耐受反应,对于心肌缺血、脑卒中、高血压、循环血管再通和组织移植等可能会具有广阔的临床应用前景。同时,CGRP在偏头痛的发病机制中起重要作用,利用CGRP受体拮抗剂可以有效治疗偏头痛。
神经肽Y过度释放引起心肌缺血和脑血管痉挛
神经肽Y(neuropeptide Y,NPY)由36个氨基酸残基组成,其结构与胰多肽(YY肽)相似,同属一个肽类家族,由于其分子中富含酪氨酸,故又称为神经肽酪氨酸。NPY基因由7200bp组成,含4个外显子和5个内含子,它可以转录、表达成97个氨基酸残基组成的NPY前体,贮存于神经纤维的分泌囊泡内,释放时再经酶解,分解成有活性的NPY。NPY主要分布在中枢和外周神经系统中,心血管系统亦有丰富的NPY神经纤维,NPY由心脏交感神经末梢随同儿茶酚胺一起分泌。在外周神经系统,它主要与去甲肾上腺素(NE)共存于交感神经中,并由交感神经末梢释放。心脏的NPY含量最多,每克心肌含150pmo1。NPY是冠脉循环的调节物,NPY的过度释放可能是引起心肌缺血、脑血管痉挛的重要原因,而且阻碍心脏侧支循环的建立和作用。这种作用不能被阿托品、肾上腺能α和β受体阻断剂、5-羟色胺拮抗剂以及前列腺素抑制剂所遏制,但应用Ca2+拮抗剂或降低细胞外Ca2+浓度时,则可显著抑制NPY的缩血管效应。因此,NPY的缩血管作用依赖于细胞外Ca2+浓度。
除了对血管的直接作用之外,NPY还可加强其他缩血管物质(去甲肾上腺素、组胺等)的反应性和抑制舒张血管物质(腺苷、乙酰胆碱和β受体阻断剂等)的舒血管效应。NPY还是迷走神经的调节因子,静脉注射NPY可长时间抑制迷走神经兴奋所引起的心动过缓。其作用机制可能与腺苷环化酶和G-蛋白激活有关,并通过与NPY1和NPY2受体结合而影响血管张力和心脏功能。
此外,NPY在高血压发病中亦可能具有一定意义。自发性高血压大鼠SHR下丘脑和脑干NPY的含量明显增高。肾上腺髓质嗜铬细胞瘤高血压的患者,血中NPY的水平亦明显高于正常人。因此,寻求NPY的拮抗剂和阻断剂,将可能为动脉粥样硬化、冠心病、脑梗死和高血压的治疗开拓一条新的防治途径。
速激肽扩张血管
速激肽(tachykinin,TKN)是一类单链多肽,其肽类家族含有20多个成员,其中主要有P物质(SP)、K物质(SK)、神经激肽B(neurokinin B,NKB)和神经肽K(neuropeptide K,NPK)等。SP和SK主要存在于中枢神经系统和消化系统。近年发现在心血管系统亦广泛分布,其神经纤维主要来源于心脏的星状神经节。速激肽神经纤维在冠状动脉分布密集,此外,还支配主动脉、肺动脉、肾和脑血管。速激肽具有3种类型的受体(NK-1、NK-2和NK-3),其相应的内源性配基分别为SP、SK和NKB。常与CGRP共存于同一神经纤维的同一分泌囊泡内。这类受体可能与G蛋白耦联,以磷酸肌醇为第二信使,发挥其生物学效应。但糖皮质激素与受体结合后可减少速激肽受体的表达,其作用是通过减缓速激肽NK-2受体基因转录的速度而实现的。
速激肽具有舒张冠状动脉、脑动脉、肺动脉和外周血管的作用。可对抗去甲肾上腺激素所引起的缩血管作用。一方面,SP舒张冠状动脉的作用大于SK;另一方面,SP的舒血管作用可能依赖于内皮细胞,去除血管内皮细胞后,其舒血管作用明显减弱。应用SP或SK灌流冠状动脉,可明显降低冠状动脉灌流压,增加A-V氧分压差,提高心脏排血量。静脉注射速激肽可降低平均动脉压、左室收缩末期压和最大收缩速率。但是,应用SK灌流离体心脏,则可增加心肌收缩力,产生正性变时和变力效应,而同样剂量的SP则无此作用。此外,速激肽增加血管通透性,促进血管平滑肌细胞增殖和心房肌细胞释放ANP。
血管活性肠肽增加心脑血流量并降低外周阻力
血管活性肠肽(VIP)前体是在神经末梢贮存的主要形式,由170个氨基酸残基组成,其中81~108位片段为富含组氨酸多肽(PHM),125~152位片段为VIP。VIP的C端片段(18~28)是维持其生物活性所必需的,肽链越长,活性亦越高。近年来发现,N端片段(1~10)亦有生物活性,但逊于VIP18~28。由于VIP1~l0和VIP18~28的组成不同,而又都具有生物活性,因此推测VIP可能具有两个活性“信息”片段。VIP主要分布于中枢神经系统和胃肠道内。作为神经递质,其神经纤维广泛分布于心血管系统。血浆中的VIP浓度仅为18pmol/L,半衰期约1分钟,主要在肝和肾灭活。
VIP是内源性血管舒张剂,应用生理剂量的VIP(0.3μg/ kg),可使心、脑、肾、肺、消化腺、汗腺和骨骼肌的血管舒张,增加心、脑血流量,降低外周阻力,使血压特别是舒张压降低。此外,它还有增加心率、心肌收缩力和血管通透性的作用(以cAMP或以cGMP为介导)。VIP的舒血管作用与血管紧张度有关,血管紧张性越高,其舒张作用越强。VIP舒张血管的作用不受肾上腺素能和胆碱能受体阻断剂的影响,是一种非胆碱、非肾上腺素能的血管舒张剂。其作用不及异丙基肾上腺素和乙酰胆碱,但作用时间长。一般认为前列腺素合成抑制剂不能阻滞VIP的舒血管作用。大剂量吲哚美辛(消炎痛)可部分抑制VIP的舒血管效应。VIP的舒血管效应在肺动静脉和主动脉是内皮细胞依赖性的,而在另一些血管如冠状动脉和脑血管则是非内皮细胞依赖性的。
失血性、内毒素性和内脏缺血性休克,循环血液中的VIP成倍增加,其中肠系膜静脉的VIP远高于体循环和肺循环,这可能与胃肠道大量释放VIP有关。由于VIP可以舒张心、脑血管,增加血流量,具有正性心脏变力作用,因此在休克的早期具有代偿意义。但在休克晚期,VIP强烈舒张内脏血管,增加毛细血管的通透性,使血压呈进行性下降,促进休克时小肠的再灌注损伤。
VIP肿瘤可导致低钾、胃酸缺乏和水泻综合征。
缓激肽增强血管通透性并降低血压
缓激肽(bradykinin,BK)是由激肽释放酶(kallikrein)作用于激肽原(kininogen)所产生的一类局部激素,主要有缓激肽、胰缓激肽和甲胰缓激肽3种。它们是不同的激肽释放酶作用下的不同产物,但其结构基本相同。激肽释放酶、激肽原、激肽和激肽酶组成激肽释放酶-激肽系统(kallikrein-kinin system),见下图。激肽原在激肽释放酶作用下产生激肽。但由于体内广泛分布有激肽酶,使激肽迅速降解,因此激肽是一种局部激素。激肽酶Ⅰ和Ⅱ都是羧基肽酶(分别为28kD 和12kD的糖蛋白)。激肽有Bl和B2两种受体,在心血管系统主要为B2受体。
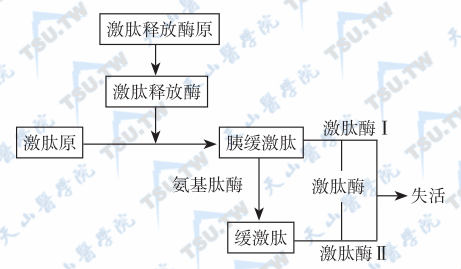
激肽释放酶-激肽系统
缓激肽和胰缓激肽是体内最强的血管舒张物质之一。静脉注射BK可引起全身小动脉舒张、血管通透性增强,血压降低。BK与内皮细胞上BK受体结合,释放内皮舒张因子,再作用于血管平滑肌而使血管舒张;BK对冠状血管具有双重作用,BK通过释放内皮舒张因子,使冠状动脉舒张;而BK的代谢产物——去精氨酸BK直接作用于冠状血管平滑肌,使血管收缩。BK不仅是肾脏血流的主要调节因子,而且还具有强大利钠利尿效应。这种作用可被其抗体或拮抗剂所遏制。人体内血浆激肽水平与肾素活性呈正相关,注射激肽可促进肾素的释放。激肽与高血压的发病可能有关。流行病学调查发现非洲人较白种人高血压的发病率高,其尿中激肽释放酶水平较白种人低。缓激肽与心绞痛和心肌梗死亦有密切关系。BK是心绞痛的主要致病因子,在心绞痛和急性心肌梗死(AMI)时,血BK和激肽释放酶活性均明显增加。激肽释放酶、激肽原亦可激活凝血机制,使局部血流处于高凝状态,从而可加速血管的梗死。在心肌梗死的局部,激肽的含量明显高于非梗死区。心源性休克时,血浆激肽亦显著升高,其升高幅度与血压、左室收缩末期压(LVESP)和心排血量呈负相关。
内皮素促进血管平滑肌细胞增生
内皮素原(preproendothelin)含203个氨基酸残基,经肽酶水解后形成ET原,再经转化酶的作用形成ET。ET不仅存在于主动脉内皮细胞,其他血管的内皮细胞亦含有ET。人类有3种ET基因,分别表达ET-1、ET-2和ET-3。其生物活性以ET-1最强,ET-3最弱。ET的C端和其环状结构对维持其活性十分重要,若去除21位的色氨酸,或用D-色氨酸代替,其活性明显降低;若用内肽酶在第9位水解赖氨酸或将ET分子中赖氨酸碳胺基甲酰化(carboxamidomethylation),其活性降低99.5%。
在生理状态下,血浆ET的浓度极低(<5.0pg/ml),降解慢。血管平滑肌细胞和肺是ET降解的主要场所。转化生长因子、缺血、缺氧、凝血酶和肾上腺素等可以刺激前ET原基因的转录,AVP和AT-2可促进ET的释放。体内许多组织器官都含有ET受体,主要分布在肺、肾和肾上腺,其次为心脏、主动脉和胃肠道。应用125I-ET的放射自显影证明,在肾小球和血管平滑肌细胞上具有密集的ET结合位点。ET与膜受体的结合不受P物质、CGRP和NPY等的影响。
对心肌的作用
ET是体内最强的心脏收缩因子。使用外源性ET可导致冠状动脉痉挛、心律失常、心排出量减少和组织蛋白质漏出,心肌细胞内钙聚积等。ET直接增加心肌收缩力,且不受β-和α-肾上腺素能、组胺能神经和5-羟色胺能等受体阻滞剂或吲哚美辛(消炎痛)的影响。
对血管平滑肌的作用
静脉对ET比动脉更敏感。在动脉中,冠状动脉对ET较敏感。ET引起的血管收缩对α-肾上腺素能、H1-组胺能、5-羟色胺能、毒蕈碱样受体拮抗剂以及环氧化酶、脂氧合酶抑制剂均有拮抗作用。ET-1参与高血压发病过程,它可促进血管平滑肌细胞的增生与肥大。在细胞凋亡晚期,ET-1抑制22碳六烯酸(docosahexaenoic,DHA)诱导的caspase 3(死亡信号通路的蛋白裂解关键酶)活性及其DNA分裂,从而抑制DHA诱导的细胞凋亡,血管平滑肌细胞的凋亡减少。
与心脑肾肺疾病的关系
ET的大量释放被认为是机体在某些疾病状态下的一种内源性致病因素,与心、脑、肾和肺等疾病的发病过程有关。ET使冠状血管收缩,引起冠状血管的痉挛甚至猝死。ET-1的远期效应是使心肌肥厚和心肌细胞损伤,心肌梗死患者或急慢性心衰时血浆ET-1显著增加;原发性高血压患者,血管对ET敏感性增加,血浆ET轻度升高;ET能引起脑血管强烈收缩,脑卒中患者血浆ET显著增加;ET可使支气管和肺动脉强烈持久收缩,引起呼吸困难。此外,ET还有致消化性溃疡作用,可引起胃出血。感染性休克者的血浆ET较正常人高3~5倍,故认为ET可能是体内的一种休克因子;ET对肾血管具有强烈收缩作用。
内源性洋地黄素与高血压有关
洋地黄类制剂是治疗心功能不全最常用的药物。心脏含有类似洋地黄作用的利钠因子,可抑制Na+/K+-ATP酶,在放射免疫测定中与地高辛抗体有交叉反应[内源性类洋地黄因子或内洋地黄素(endodigin,endigen)或内地高辛素(endoxin)]。内源性洋地黄素(endogenous digitalis-like factor)具有3种分子形式(5000u、500u和230u),在血浆中主要以小分子形式存在。不同分子形式的内源性洋地黄素均有3种共同的生物学作用:①抑制Na+/K+-ATP酶。②特异性与哇巴因受体结合。③可与地高辛抗体发生交叉反应。
正常人和高血压患者血中均可测到内洋地黄素。但高血压患者血中内洋地黄素的水平远超过正常人,其含量与高血压的水平亦呈正相关。在高血压患者中,低肾素性高血压患者血中内洋地黄素的水平又高于正常肾素性高血压患者。高血压患者经ACEI治疗后,随着血压的降低,血浆内源性洋地黄素亦降低。内源性洋地黄素活性增高,抑制Na+/K+-ATP酶,阻遏细胞内Na+的外流,细胞内Na+潴留,增强心肌收缩力和缩血管反应性,可能是原发性高血压的重要发病机制。慢性心功能不全、醛固酮增多症、慢性肾功能不全、肝硬化和甲状腺功能亢进等疾病时,血浆和尿内内洋地黄素亦明显升高。
肾素-血管紧张素调节冠脉循环和心肌收缩
RAA中的AT-2在血压的调节中起着中心作用。AT-2是一种有效的血管收缩因子,它直接作用于血管平滑肌,增加血管平滑肌的一些调节因子的受体密度和敏感性,如FGF、TGFβ-1、PDGF和IGF。AT-2作用于交感神经系统影响心肌的收缩力和心率,激活巨噬细胞、增加血小板聚集、刺激血浆纤溶酶激活抑制因子-1(PAI-1),直接引起内皮细胞功能障碍。AT-2促进动脉粥样硬化的机制在于它能抑制细胞凋亡、增强氧化应激、促使白细胞黏附和迁移以及促进血栓形成。ACEI能有效地抑制AT-2的合成,有利于延缓或阻滞疾病的进程。这在糖尿病肾病、心肌梗死或心衰的临床应用中已得到证实。
心脏内具有RAA全部的组分,具有一个独立于肾脏的RAA。心内RAA受一些体液和神经递质的调节。低钠负荷促进心内肾素和AT原的表达,增加其mRNA水平,提高心内肾素的含量。异丙肾上腺素和雄激素亦可促进心内肾素的合成,而非特异的血管舒张剂肼屈嗪(肼苯达嗪,hydralazine)亦可增加心内RAA活性。钙拮抗剂则降低其活性。甲基多巴和ACEI对心内肾素无明显影响。
心内RAA的作用机制尚不十分明了。研究证明,局部合成的AT-2在心血管的重塑过程中起着关键性作用,它以旁分泌和自分泌方式影响血管的结构和功能,调节冠脉循环,引起冠脉血管收缩;增加心肌收缩力,促进心内交感神经末梢释放儿茶酚胺;通过心肌细胞核上的AT受体,促进心肌细胞蛋白质的合成,刺激心肌细胞增生,而致心肌肥厚;参与心肌细胞间信息交流。此外,还加重和诱发心肌缺血或再灌注损伤,诱发心肌缺血所引起的室性心律失常。应用ACEI可以阻止上述反应,减少心肌缺血所引起的损伤面积,降低乳酸脱氢酶和肌酸激酶的活性,减少乳酸的产生,改善心脏的血流动力学,增加左室收缩力,减少氧耗,增加冠脉血流量,提高心肌组织内的糖原、ATP和磷酸肌酸,具有显著的心肌保护作用。该旁路产生的AT-2可被组织蛋白酶G(cathepsin G)、抑凝乳蛋白酶素(chymostatin)、敏感性AT-2生成酶和凝乳酶(chymase)等降解。研究发现,AT-2的缩血管作用是通过刺激AT-2的1型受体(AT-1受体)来实现的,并发现AT-1受体基因在3’转录区+A1166C突变与高血压和冠心病有关。
