
β珠蛋白生成障碍性贫血(β地中海贫血)是由于β珠蛋白基因突变导致β珠蛋白链合成不足而引起的溶血性贫血。本病广泛流行于地中海流域、中东、非洲及中国南部等,在我国广东、广西、四川、香港、台湾北部、云南、贵州、海南、福建、湖南、湖北也是高发区。β地中海贫血是危害最为严重的血红蛋白病,也是世界上最常见的遗传性疾病之一。大约占世界人口的3%,约1. 5亿人群携带β地中海贫血基因。
病因
正常人自父母双方各继承一个正常β珠蛋白基因,合成正常量的β珠蛋白链。若自父母继承了异常β珠蛋白基因,则可导致本病。由于β珠蛋白基因突变部位和类型不同,对β珠蛋白合成抑制的程度也不同,常将β地中海贫血分为两种类型,β珠蛋白完全不能合成者称为β0地中海贫血;β珠蛋白尚能合成但合成量不足者称为β+地中海贫血。
发病机制
人类β珠蛋白基因位于11号染色体上,已知将近有150种基因突变可导致β地中海贫血。由于β珠蛋白基因发生突变,导致β珠蛋白基因的转录、前体mRNA的加工、mRNA的翻译及β珠蛋白链的完整性发生障碍,导致β珠蛋白链的合成不足或完全不能合成,引起α珠蛋白链与非α珠蛋白链的合成比例不平衡,影响正常的血红蛋白(HbA)的合成。此外,由于α珠蛋白链的相对过剩,剩余的α珠蛋白链在红细胞内形成包涵体,导致红细胞膜的氧化损伤,造成红细胞破坏及骨髓的无效造血,临床上引起贫血、黄疸、脾大、骨髓腔扩大引起的地中海贫血外貌等症状及体征,这是β地中海贫血主要病理基础。
β地中海贫血的HbA(α2β2)减少而HbF(α2γ2)、HbA2(α2δ2)增多。HbF和HbA2增多的归因于:①对红母细胞前体的选择,有利于产生更多的HbF和HbA2;②红母细胞的扩增有利于γ-链和δ-链的产生。
临床表现
临床上按其贫血严重程度分为轻型β地中海贫血、中间型β地中海贫血和重型β地中海贫血。轻型β地中海贫血为杂合子β地中海贫血,多数患者没有任何症状,也无贫血;少数有轻度贫血,面色较差,常感疲乏无力,但生长发育正常,骨骼无畸形。贫血可因感染、妊娠等情况加重,也可并发缺铁性贫血,脾可轻度肿大。重型β地中海贫血又称Cooley贫血,为纯合子β地中海贫血,β珠蛋白链合成完全被抑制(β0地中海贫血),初生时与正常婴儿无异,但出生后3~6个月,随着γ珠蛋白基因表达逐渐关闭,由于β珠蛋白基因缺陷,患者开始出现临床症状,且呈进行性加重,须定期输血维持生命。早期症状如食欲不佳、喂养困难、腹泻、激惹、发育缓慢、体重不增,面色逐渐苍白,肝脾特别是脾进行性肿大,腹部逐渐膨大。三四岁起体征越来越明显,贫血进行性加重,巩膜黄染、生长发育迟缓、身体矮小、肌肉无力,骨骼变形,头颅增大,额部、顶部、枕部隆起,颧骨隆起,鼻梁塌陷,上颌及牙齿前突,形成典型的“地中海贫血外貌”。
巨脾可因脾功能亢进而引起粒细胞和血小板减少,时常有感染、发热、鼻出血等。长期多次输血常引起继发性血色病,免疫力低下、反复感染、心肌损害,常使多数患儿夭折。如能活到10多岁则常伴性幼稚征,出现第二性征不发育、肾上腺功能不全等症状。中间型β地中海贫血是指不依赖输血,临床表现介于重型与轻型β地中海贫血之间的β地中海贫血患者。从遗传因素分析,至少包括:
- 轻型的纯合子β地中海贫血(β+地中海贫血);
- 贫血和脾大比较明显的杂合子β地中海贫血;
- 纯合子β地中海贫血复合α地中海贫血;
- 纯合子β地中海贫血复合γ珠蛋白基因启动子突变;
- β地中海贫血复合异常血红蛋白,如HbC、HbE、HbS等;
- β地中海贫血复合δβ地中海贫血;
- β地中海贫血复合Hb Lepore。
实验室检查
一、血象:轻型β地中海贫血Hb一般在80g/L以上,重型β地中海贫血Hb在一般50g/L以下,需定期输血维持生命,MCV、MCH、MCHC明显降低。网织红细胞比率常增高,血涂片检查见靶形红细胞增多、红细胞大小不均、异形、嗜碱性点彩明显,红细胞呈典型小细胞、低色素性。白细胞数多正常,血小板数常增高,脾功能亢进时白细胞、血小板数减少。
二、骨髓象:呈溶血性贫血骨髓象,红细胞增生显著,铁染色阳性,铁粒幼细胞增多。
三、血红蛋白分析:轻型β地中海贫血HbA2显著增高,范围3. 5%~7%,平均5%。HbF可以正常,部分病例可以轻度增高,一般不超过5%。重型β地中海贫血HbF增高明显,可达60%以上,有些患者HbF为10%~30%。HbA2多正常,变化较大,范围在1. 4%~4. 1%。
四、铁代谢检查:轻型β地中海贫血患者的血清铁、铁饱和度、血清铁蛋白浓度多数正常,合并缺铁时上述指标可降低。中间型及重型β地中海贫血患者的血清铁、铁饱和度、血清铁蛋白浓度常增高,重型β地中海贫血上述指标增高更为显著,其中血清铁蛋白浓度常>2000μg/L。
五、X线检查:重型β地中海贫血患者骨髓长期和显著增生,使骨髓腔增宽、骨皮质变薄,颅骨板障增宽。颅骨X射线片上常能看到骨皮质间的髓梁有垂直条纹,呈典型短发状变化,如“头发直立”“太阳光线”状。短骨由于骨小梁变薄而形成花边或嵌花样间隔,以指骨及掌骨出现较早,长骨骨皮质变薄髓腔增宽,以股骨远端较明显。偶在胸腔内或脊柱旁可以见到大小不等的髓外造血灶。
诊断
纯合子β地中海贫血的临床和血液学表现很典型,诊断并不困难。对于进行性严重贫血的患儿,有脾大,外周血片显示红细胞大小不均、有靶形红细胞,红细胞渗透脆性降低,HbF含量显著增高,大多可以确诊。家族史和籍贯对诊断有重要意义,必要时做颅骨X线检查及血红蛋白分析,疑似病例需做基因诊断。轻型及无症状β地中海贫血的诊断依据:①低色素性贫血。②外周血涂片可见靶形红细胞。③红细胞渗透脆性减低。④HbF正常或轻度增多,HbA2轻度增多,血红蛋白电泳无其他异常血红蛋白。⑤家族调查对诊断很有价值,患者的父母中至少一人有杂合子β地中海贫血证据,也可两人都有。杂合子β地中海贫血需与缺铁性贫血、巨幼细胞贫血相鉴别,纯合子β地中海贫血需与新生儿黄疸、再生障碍性贫血等相鉴别。
治疗
轻型β地中海贫血无需治疗,中间型及重型β地中海贫血采用以下措施治疗:
输血
目的是维持患儿的正常血红蛋白水平,以防慢性血氧不足,减轻代偿性骨髓增生,减少肠道对铁的吸收。重型β地中海贫血目前主张采用高输血法维持患者Hb在100~120g/L。一般每3~4周输血1次,由于患者每周Hb下降约10g/L,4周则下降40g/L,而提升Hb 10g/L,每千克体重需3ml洗涤红细胞或浓缩红细胞(相当于5ml的全血),因此每千克体重需输12ml红细胞(相当于20ml全血)来使Hb上升40g/L。如果患儿体重20kg,则需240ml红细胞(相当于400ml全血)来使Hb上升40g/L。在输血过程中应用洗涤红细胞或用过滤器去除白细胞后的浓缩红细胞,可减少输血后的过敏反应及肝炎、艾滋病、巨细胞病毒等传染性疾病的发生。中间型β地中海贫血大多数平时能维持Hb在75g/L以上,无需依赖长期规则输血。只有在临床症状表现如重型β地中海贫血时,才需长期规则输血,若感染后,暂时的Hb下降,输血后可回升,对孕妊期间的中间型β地中海贫血患者,需规则输血。
铁螯合剂治疗
长期反复输血及骨髓红系细胞造血过盛、肠道吸收铁增加,使体内铁负荷过重,过多的铁沉积于心肌、肝、胰、脑等全身器官,引起血色病。临床上常有面色铁青、心力衰竭、肝硬化、生长发育障碍、糖尿病等。其中心力衰竭是引起患者死亡的主要原因,故应用铁螯合剂进行去铁治疗对β地中海贫血十分重要,但过早使用铁螯合剂可影响小儿骨骼生长,故常须对体内铁负荷进行评估,最普遍使用的评估方法是测定血清铁蛋白浓度。但由于血清铁蛋白易受溶血、炎症、肝病等因素的影响,故数值不够准确;且铁主要沉积在一些高水平表达转铁蛋白受体的组织器官,如肝、心脏及内分泌器官,血清铁蛋白仅反映了1%的贮存铁水平。活检获得肝铁质浓度被认为是评价体内铁负荷最准确而敏感的指标,但肝穿刺是侵入性检查,具有一定的危险性,在一般的医疗单位,尤其幼年患儿难于进行。目前磁共振检查(T2*)及超导量子干涉装置(SQUID)已被用于检测组织贮存铁。
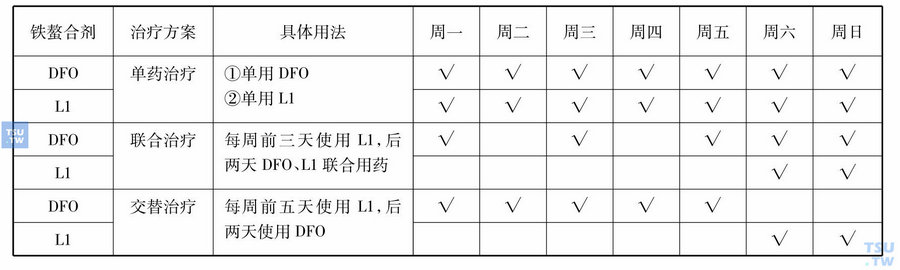
地中海贫血国际联合会公布的2004年地中海贫血的临床治疗指南中表明,接受输血10~20单位红细胞或血清铁蛋白浓度在1000μg/L以上时应开始应用去铁治疗。目前可选择的铁螯合剂有:去铁胺(deferoxamine,DFO),去铁酮(deferiprone,L1),及地拉罗司(deferasirox,Exjade)。DFO为六配基铁螯合剂,是目前使用最广泛而且效果最好的铁螯合剂,也是唯一被批准在美国和加拿大使用的铁螯合剂,已被广泛地使用了20多年,需要静脉或皮下缓慢注射,每天剂量20~50mg/kg维持8~12小时,每周5~6天。L1为二配基口服铁螯合剂,有效剂量为75mg/(kg•d)。与DFO相比,其优点有:①口服螯合剂;②可进入细胞,清除细胞内沉积的铁离子;③去除心脏铁沉积效果优于DFO。Exjade是一种新型的口服三配基铁螯合剂,每天剂量20~30mg/kg。研究结果显示,当Exjade服用剂量≥30mg/(kg•d)时,可获得显著的去铁效果;剂量<20mg/(kg•d)时,Exjade去铁效果不理想;当剂量≥20~<30mg(kg•d)时,Exjade可保持铁负荷的平衡。因Exjade具有良好的耐受性和动员组织铁的功效,可促进铁排泄,每日一次的服用方法易于被大多数患者接受,其治疗依从性好。铁螯合剂使用方法上可选择单药治疗或两种铁螯合剂联合或交替用药,用法可见表:
铁螯合剂使用方法
脾切除及脾动脉栓塞
对巨脾或(及)脾功能亢进者可行脾切除术或脾动脉栓塞术,以减轻溶血。切脾指征:①脾大6cm以上或脾功能亢进;②每年输血量超过200~250ml/kg红细胞者;③5岁以上(5岁以前小儿机体免疫功能发育未完善,术后常并发严重感染)。脾切后患者输血量减少,红细胞寿命延长、贫血症状改善,目前认为HbH病、中间型β-地贫、β-地贫复合HbE病患者脾切效果好,脾切后因免疫功能减低容易合并感染,同时血小板明显增高,易导致血栓栓塞,肝含铁血红素沉积加重并明显增大,其他器官亦受累。脾切后因立即给予抗生素预防感染1~2个月。血小板>800×109/L者应给予阿司匹林、双嘧达莫等抗凝治疗。
抗氧化剂
如维生素E 50mg/d,维生素C 100~200mg/d;阿魏酸钠(当归的成分之一),剂量为150~300mg/d等能稳定红细胞膜,减轻溶血。
γ珠蛋白基因活化剂
如羟基脲(hydroxycarbamide)剂量为25~50mg/(kg•d),5-氮胞苷(azacytidine,5-Aza)、白消安(busulfan)、丁酸钠类等药物,能活化γ珠蛋白基因的表达,增加γ珠蛋白链的合成,增加HbF的合成,改善贫血症状。该类药物对中间型β地中海贫血效果较好,但对重型β地中海贫血效果较差。
造血干细胞移植
异基因骨髓移植、外周血干细胞移植及脐带血移植是目前根治重型β地中海贫血的唯一方法。对有HLA相合同胞供体的重型β地中海贫血患者来说应作为首选治疗。骨髓移植效果与患者年龄、身体状况、预处理方案、供者来源、HLA相合程度及对并发症的处理等因素密切相关。根据地贫患者是否肝大,肝纤维化,是否规则应用铁螯合剂三种危险因素,将患者分为三级:一级为无上述3种危险因素;二级有1~2种危险因素;三级有3种危险因素。最近20年来,由于预防措施的改进、移植相关并发症的有效控制、新的预处理方案的应用,HLA相合的同胞移植获得了相当的成功。目前上述三级患者获得HLA配型相合供者的骨髓移植后,无病生存率分别为87%、85%和80%。对于没有HLA相合同胞供体的重型β地中海贫血患者而言,可选择无关供者造血干细胞移植根治疾病。
预防
重型β地中海贫血目前总体预后较差,多数于学龄前因继发感染、全身及心力衰竭而死亡,总体上临床控制的效果仍不理想。因此,预防控制显得尤为重要。预防控制的主要措施包括通过社区筛查、遗传咨询和产前诊断手段控制重型β地中海贫血患儿的出生。对于有重型地贫患儿出生史、夫妻均为地贫携带者的高危孕妇应严格进行产前诊断。产前诊断包括取胎儿绒毛、羊水及胎儿脐带血作基因分析。其中以早期绒毛为首选,取胎儿绒毛以孕8~12周为最佳时间;若错过采集绒毛的时机,可于孕16~24周采集羊水,并经培养去除母血细胞后提取胎儿DNA进行基因分析;经胎儿脐静脉穿刺取血样提取胎儿DNA也可以用于产前诊断,一般在孕20~26周进行。但上述均为侵入性产前诊断途径,存在一定的风险性,近年来逐渐发展了一种非损伤性、非侵入性产前诊断方法,即从孕妇外周血中提取胎儿DNA用于基因分析。
国内龙兴江等首次利用孕妇外周血浆中的游离胎儿DNA以及母源性游离DNA 在PCR反应中扩增效率的不同,对巴氏水肿胎儿进行无创性产前诊断,方法安全、可靠。在优化实验条件的前提下,该方法有望用于α0基因携带者和巴氏水肿胎儿的无创性产前基因诊断。产前诊断是预防与监控的结果,具有很强的社会意义和实用价值,特别是在地贫高发区是控制重型地贫患儿出生,预防地贫的关键。
