
PIDs包括特异性免疫缺陷和非特异免疫缺陷,本节在介绍重要的常见的免疫缺陷病的同时,摘要叙述几种新近确定的罕见的免疫缺陷病。
特异性免疫缺陷(specific immunodeficiency).绝大多数PID由特异性免疫组分缺陷所引起。这种特异性缺陷影响到T细胞和B细胞的发育、活化和相互间协同作用。从疾病的分类中可以看到,有些免疫缺陷的临床表型以体液免疫缺陷为特征,但实质是免疫细胞或分子间相互关系缺陷的结果。
T细胞和B细胞联合免疫缺陷(combined T and B cell immunodeficiencies)。严重联合免疫缺陷病(severe combined immunodeficiency,SCID)分为两组,T-B-SCID和T-B+SCID。其中,X连锁T-B+SCID占多数,主要是γc链的突变;T-B-SCID则源自细胞间激酶JAK3(与γc结合)的突变。在常染色体隐性遗传的SCID中,腺苷脱胺酶缺乏症约占50%。部分SCID的遗传学和分子基础已被阐明。一些SCID患者,在新生儿期,由于母体淋巴细胞植入,可出现类似于移植物抗宿主病(GVHD)的症状(Omenn综合征)。在输血或血制品时,因接种了同种异体淋巴细胞,可发生致死性的GVHD。部分SCID通过骨髓移植获得治愈。
1. RAG-1/-2缺陷
本病系常染色体隐性遗传(11p12-13),因淋巴系统特异性重组激活基因(recombination activating gene,RAG)突变(RAG-1或RAG-2),导致T-B-SCID或Omenn综合征。RAG-1 和RAG-2参与Ig和TCR基因V-D-J的重排,是DNA重组形成Ig和TCR基因之前的剪切双链DNA所必需的,在T、B细胞抗原受体的形成中起着极为重要的作用。
T-B-SCID患儿的临床表现与其他类型SCID相似,外周血除NK细胞升高外,T细胞和(或)B细胞计数很低,不能产生免疫球蛋白。Omenn综合征患儿胸腺完全缺乏T细胞,淋巴器官萎缩,表现有红皮病、鳞屑样皮炎和脱发,嗜酸细胞增多,肝脾和淋巴结肿大,淋巴结无淋巴细胞,代之以巨噬细胞;血清IgE增高,外周血淋巴细胞显著增加,酷似移植物抗宿主病(GVHD)。由外周血T淋巴细胞能找到表达激活标记物的CD25和MHCⅡ类分子。诊断应排除因母体T细胞输注或接受血液制品导致的GVHD。
2.腺苷脱胺酶缺乏症(adenosine deaminase,ADA)
10%~20%的SCID由ADA缺乏所致,占常染色体隐性遗传SCID的50%,其发病率为每20万新生儿中有1例。ADA基因定位于20q13. 2-13. 11。多数ADA基因突变为CpG二核苷酸C→T点突变,整个基因或部分基因缺失仅见于少数病例。在嘌呤代谢的补救途径中,ADA不可逆地催化腺苷和2’-脱氧腺苷分别成为肌苷和2’-脱氧肌苷。
ADA缺乏使脱氧三磷酸腺苷(dATP)积累,后者抑制核糖核苷还原酶,进而抑制细胞DNA的合成。发育初期,向T、B淋巴细胞转化的骨髓干细胞不能降解dATP(相对缺乏5’核苷酶),导致T细胞和B细胞数量减少和功能减低,产生联合免疫缺陷病。患儿出生不久出现“三感染”(口腔白色念珠菌、顽固性腹泻和肺炎),均有淋巴细胞和免疫球蛋白减少。约10%的患儿初期免疫球蛋白水平正常,但仍会逐渐减少。部分性ADA缺陷或嵌合体者无任何临床表现。可通过测定红细胞或淋巴细胞的ADA活性做出临床诊断。基因分析有助于家系调查。
3. X连锁SCID(X-SC ID)
本型约占SCID的半数。突变基因位点在Xq13. 1-13. 3。因编码的IL2受体γ(γc)链基因突变,导致IL2、4、7、9和15受体共用γ链异常,不能刺激未成熟的胸腺细胞生长,导致T细胞分化障碍。患儿胸腺发育不良,外周淋巴结极小,且无淋巴细胞。尽管B细胞数量正常,由于不能获得T细胞的辅助,患者同样有体液免疫缺陷。当免疫球蛋白亚类发生紊乱时,则出现免疫球蛋白增高的Nazelof综合征。
4.网状组织发育不全(reticular dysgenesis)
本病为常染色体隐性遗传,临床罕见。淋巴系和髓系前体细胞成熟障碍。患儿不仅有严重的淋巴细胞减少,粒细胞和血小板也严重减少。患儿出生不久(3天~3个月)即夭折。
5.嘌呤核苷磷酸化酶缺乏(PNP)
本病为常染色体隐性遗传,PNP的基因位于14q13. 1。发病率远较ADA少。不同患者T细胞免疫缺陷程度不同,且病程缓慢,临床症状可延迟到数年之后才出现,对各种感染的易感性增高,输血后可引起弥散性致死性水痘和GVHD。
PNP与ADA一样,是嘌呤代谢的降解酶。PNP缺乏时,鸟苷转化为鸟嘌呤、肌苷转化为次黄嘌呤的通路受阻,使鸟苷、脱氧鸟苷和dGTP堆积,后者抑制核苷酸还原酶,阻断DNA的复制,进而影响淋巴细胞的生长发育。由于dGTP对T细胞的毒性程度大于B细胞,因此,PNP与ADA的免疫学异常存在程度的差别。
6. MHCⅡ类缺乏(MHC classⅡdeficiency)
本病为常染色体隐性遗传,因抗原递呈细胞(APC)表达MHCⅡ类分子障碍所致。由于MHCⅡ类分子或Ⅰ类分子缺乏时,T细胞表面似裸细胞,因而称之为裸淋巴细胞综合征(bare lymphocyte syndrome)。现已明确,RFX5(异二聚体RFX5,1q21)和CⅡTA(Ⅱ类分子转录激活基因,16p13. 1-2)基因的突变与本病密切相关。由于胸腺内和外周血APC均不能表达MHCⅡ类分子,从而影响CD4+T细胞的阳性选择,使循环中接受APC抗原递呈的CD4+T细胞减少,导致抗体合成和血清免疫球蛋白减少,细胞介导的免疫反应(CMI)缺失。除非骨髓移植,患儿常于1岁内死亡。
HLA-DRα和β链残缺表达是MHCⅡ类分子缺乏的一种少见表型,患者循环CD4+T细胞数量正常,临床预后较好。
7. MHCⅠ类缺乏/TAP2肽转运缺陷(MHC classⅠdeficiency/TAP 2 peptide transporter deficiency)
本病为常染色体隐性遗传(6p21. 3)。由TAP2基因突变导致内源性抗原肽不能有效地转移到内质网与MHCⅠ类分子凹槽结合(荷肽),导致MHCⅠ类分子表达减少。胸腺内CD8+T细胞的阳性选择受到影响,循环CD8+T细胞数量减少和功能减退。MHCⅠ类分子缺乏(裸淋巴细胞综合征Ⅰ型)的免疫缺陷程度和发生率均低于MHCⅡ类分子缺乏。患者表现为反复的、非病毒性呼吸道细菌感染(鼻窦炎)。这种对病毒易感性的减少的原因不明,可能是患者的NK细胞作用的结果。少数患者无症状。诊断此病的条件是:无法确定典型淋巴细胞Ⅰ类分子位点(如HLA-A、B和C),而Ⅱ类分子表达正常(HLA-DR、DP和DQ)。
8. CD3缺乏
因编码CD3γ和ε链的基因突变,使TCR-CD3不能共同接受抗原信息,导致辅助T细胞和CTL功能缺陷。CD3γ基因的点突变导致17位核苷酸的缺失;CD3ε基因的点突变导致TCRCD3复合物表达减少。因T细胞膜上CD3表达多变,即使在同一家族中,CD3缺乏表型也呈易变性。患者CD3γ和ε缺乏或异常,T细胞表面TCR-CD3表达明显减低。如果伴随IgG2缺陷,将影响多糖抗原特异性抗体的合成,而蛋白质抗原的抗体反应完全正常。CD8+T细胞(CTL)数显著减少,伴随功能障碍。
9. ZAP-70缺乏(CD8缺乏,T细胞激活缺陷,T cell activation deficiencies)
本病为常染色体隐性遗传性(2q12),系TCRξ相关蛋白激酶缺陷致细胞内信息传导障碍的免疫缺陷病。Roifman等首先描述(1989),临床罕见。ZAP-70是一种涉及TCR信号的酪氨酸激酶,其缺陷导致CD4+CD8+双阳性前T细胞不能分化为成熟的CD8+单阳性T细胞,而仅有成熟的CD4+T细胞。T细胞增殖反应低下,在抗CD3刺激作用下,T细胞不能诱导胞质酪氨酸磷酸化酶产生,钙离子流出减少。NK细胞数量和功能正常。多数患儿B细胞数量正常,但血清丙种球蛋白减少。目前已知至少有8例来自5个家族的SCID患儿为ZAP-70激酶缺乏症。幼儿期表现为反复感染和生长障碍,常见自身免疫性疾病。骨髓移植可获治愈。
10. T细胞信号传导途径中断(JAK3激酶突变)
系常染色体隐性遗传性(19p13. 1)。由JAK3激酶(Janus kinase 3)突变所致,约占SCID的10%。与γc突变引起的X-SCID很相似,属于T-B+NK-型。正常时,含JAK3激酶的IL-2受体γ链,在T细胞内激活信号传导通道中起重要连接单元的作用,即γc-Jak-Stat磷酸化后将信息转位至细胞核,激活She-Grb-Sos-Ras通路,诱导c-fos和c-jun转录因子基因表达。基因的突变仅使T细胞分化障碍,B细胞是正常的。T-B+SCID免疫缺陷的另一少见类型由IL-7受体α链突变引发,此时,T细胞发育障碍,而NK细胞存在。本病最佳的治疗方案是HLA配型相同的同胞间骨髓移植。
11.干扰素γ受体1/2(IFN-γR1/2)和白细胞介素12受体β1(IL12-Rβ1)突
变 本病为常染色体隐性遗传,由IFN-γ R1、IFN-γ R2、IL12-R β1和IL12缺乏引起。
(1)遗传基础:IFN-γ R1和IFN-γ R2基因(IF-NGR1和IFNGR2)突变(6q23-24),伴随IL12亚单位P40基因缺失。IFNGR1基因的转录产物为IFN-γR1或α链,基因突变方式为核苷酸突变、缺失、插入和拼接突变。IFNGR2基因突变导致IFN-γR2分子表达障碍,使细胞内信息传递阻止在STATS磷酸化水平。IL12亚单位P40基因缺失和IL-12缺乏导致IL-12活性下降,IFN-γ介导的巨噬细胞和T细胞功能障碍。
(2)免疫学特征:常规免疫功能检查并无异常。外源性IFN-γ诱导外周血单个核细胞产生IFN-α能力下降,T细胞对非结核分枝杆菌(NTM)的增殖反应正常,而产生IFN-γ不足。临床特征:由于IFN-γ是抗NTM感染和避免卡介苗(BCG)接种后感染的关键因子,因此,上述缺陷将导致患儿发生严重的分枝杆菌感染和BCG接种后播散性感染。
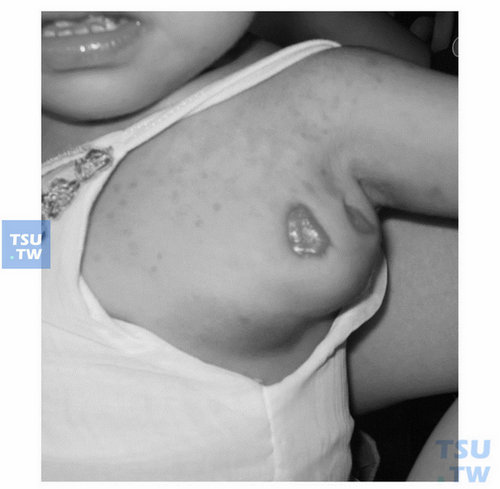
原发性免疫缺陷病合并卡介苗感染(上海复旦大学儿科医院王晓川提供)
(3)诊断:确诊有赖于IFNGR1基因缺失或IFN-γ R1分子功能缺陷的证实。自然感染NTM的临床表现与一般炎症性疾病难于鉴别,非特异性炎症性肉芽肿样的组织病理学改变和结核菌素皮肤试验结果的不统一,使临床诊断极为困难。
(4)治疗与预后:最好的治疗是分枝杆菌感染前或控制感染后的干细胞移植。自然感染NTM或接种BCG后扩散感染的患儿多数死于发病后数月至生后第二个10年内。
