
从低钾血症的正常血压患者中筛查Bartter综合征
因本病罕见,极易误诊。其诊断要点有:①血钾过低,大多在1.5~2.5mmol/L以下;②尿钾>30.0mmol/d;③代谢性碱中毒;④血浆RAA活性明显增高;⑤血压正常,且对AT-2和AVP无血压升高反应;⑥血浆前列腺素增高;⑦肾活检示肾小球球旁器的颗粒细胞明显增生及肾脏钙盐沉着;⑧临床症状典型并可排除其他因素引起的低血钾,血压不高、高血浆肾素及醛固酮,即可确诊。肾活检并非必要手段。诊断Gitelman综合征需加上低镁血症和尿镁排泄过多两项,基因诊断可明确Bartter/Gitelman综合征的病因。
Colussi等报道,利尿剂试验(diuretic test)有助于本综合征的诊断。口服氢氯噻嗪(hydrochlorothiazide)50mg(儿童用量为1mg/kg),测量服药后3小时内的利尿剂所致的氯化物清除率分数(chloride fractional clearance)。在41例病例中,Bartter综合征和假性Bartter综合征(服用利尿剂或呕吐所致)患者对氢氯噻嗪有过度反应,而Gitelman综合征患者的反应迟钝,与Bartter综合征、假性Bartter综合征或其他原因所致的低钾血症没有重叠。故可避免用复杂的基因突变分析来鉴别。
Gitelman综合征的诊断应结合症状和实验室检查的发现,典型者存在低钾血症、低镁血症、代谢性碱中毒。但是有些患者的病情很轻,或经过治疗后血清镁仅轻度降低甚至正常,从而导致误诊为Bartter综合征。另一方面,Bartter综合征患者可因饮食不足或其他继发性代谢原因而并发镁缺乏或低镁血症。因而诊断Gitelman综合征前,应首先排除非远曲小管和镁缺乏症所致的低镁血症。给患者肌肉注射硫酸镁或口服镁盐3~5天后,如果血镁转为正常,而尿镁仍无增多,则证实是镁缺乏症所致的低镁血症。反之,如果低镁血症仍存在,则提示远曲小管的镁重吸收障碍。尿钙常<0.2mmol/mmol肌酐,极少有每天超过0.5mg/ kg者。在Gitelman 综合征的诊断中,必须特别强调低镁血症和低尿钙症的重要性,如果缺乏该两种异常,一般不能诊断为Gitelman综合征;但也有反对意见(尤其是女性患者)。Gitelman综合征的特点是尿前列腺素正常,而血肾素活性和醛固酮水平仅轻度升高(Bartter综合征有明显改变)。此外,Gitelman综合征患者容易发生空泡蝶鞍综合征,其原因未明。
Bartter综合征与Gitelman综合征等病种的鉴别
Gitelman综合征
Ⅲ型Bartter综合征(CLCNKB突变所致)容易与Gitelman综合征混淆,应注意鉴别。Gitelman综合征以失盐、低钾性碱中毒、血压过低为特点,同时合并低镁血症和低尿钙症。成人发病的Bartter综合征可能以Gitelman综合征的形式出现(下表)。值得注意的是,如肾脏丢失过多Mg2+可发生低钾性碱中毒和手足搐搦,其表现类似于Gitelman综合征。Gitelman综合征还需与原发性肾性低镁血症(primary renal hypomagnesemia,无低钾血症)及继发性低镁血症(缓泻剂、慢性呕吐等)鉴别,后者的特点是尿氯化物降低。
Bartter综合征的鉴别诊断
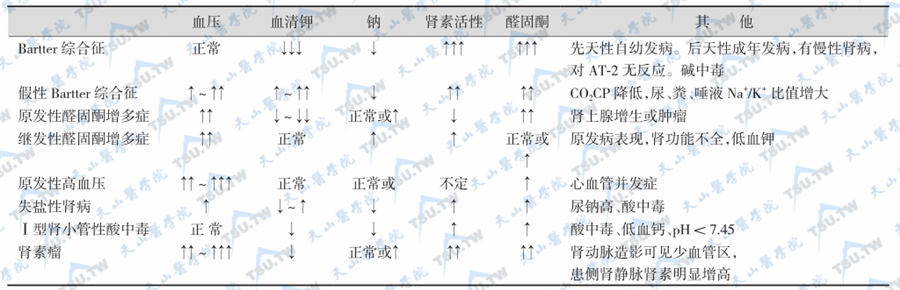
注:↓表示轻度下降,↓↓表示中度下降,↓↓↓表示显著下降;↑表示轻度升高,↑↑表示中度升高,↑↑↑表示显著升高
假性Bartter综合征
原因不明的呕吐,滥用利尿剂、缓泻剂,使用大量前列腺素、长期低氯饮食等均可产生严重、持久的低钾血症,正常血压和其他类似症状,称为假性Bartter综合征,仔细询问病史甚为关键,可疑药物引起的可停药观察。值得注意的是,有报道输注PGE亦可引起假性Bartter综合征,而真性Bartter综合征多伴有PGE升高。
原发性醛固酮增多症
有高醛固酮血症、低钾血症和代谢性碱中毒,但血压增高、肾素和AT-2均下降。
肾小管性酸中毒
有低钾血症、正常血压或肾素、醛固酮增多,但无碱中毒,相反多伴有高氯性酸中毒。
Liddle综合征
为家族性疾病,常有低钾碱血症,高血压,但无高肾素、高醛固酮血症,应用氨苯蝶啶可明显降低血压,从而与Bartter综合征鉴别。
胱氨酸病
胱氨酸病(cystinosis)表现为低钾-低氯性碱中毒,伴佝偻病和肾功能障碍,但近曲小管功能正常,角膜检查可发现胱氨酸结晶(cystine crystals)沉着。多数Ⅰ型同型胱氨酸尿症患者的生长和发育迟缓,伴有近视与虹膜震颤、青光眼、葡萄肿,白内障、视网膜剥离、视神经萎缩;骨骼系统异常表现与Marfan综合征患者相似,血清甲硫氨酸和同型胱氨酸(同型半胱氨酸)增高。
高PGE血症综合征
经典型Bartter综合征(亦包括Gitelman综合征)必须与高PGE综合征鉴别。高PGE综合征是由于肾脏ROMK1(KIR1.1)基因的失活性突变引起的产前型Bartter综合征的特殊类型,可呈家族性或散发性发病。从目前报道的病例看,以KIR1.1a的N124K突变为多见,导致PGE2的过度生成和羊水过多。但在同一家族中,可能同时存在Bartter综合征和Gitelman综合征两种类型的病例。
Dent病
Dent病(Dent disease)属于遗传性肾小管病(hereditary renal tubular disorders)的一种类型,由CLCN5突变引起的近端肾小管病,X-性连锁隐性遗传,其特点是大量排出低分子量蛋白尿、高钙尿症、肾钙盐沉着和肾石病,并缓慢进展为肾衰竭。有时,可伴有磷利尿、氨基酸尿、葡萄糖尿、尿钾增多、尿酸化功能障碍和佝偻病/骨质软化症。因此,Dent病是Fanconi综合征中的一种亚型。现认为,X-性连锁隐性遗传性肾石病伴肾衰竭、X-性连锁隐性遗传性低磷血症性佝偻病和特发性低磷血症性佝偻病均系Dent病谱中的不同类型。
SeSAME/EAST综合征
感觉神经性耳聋-共济失调-智力障碍和电解质失衡/癫痫-共济失调感觉神经性耳聋和肾小管病综合征[sensorineural deafness,ataxia,mental retardation,and electrolyte imbalance(SeSAME)/epilepsy,ataxia,sensorineural deafness,and renal tubulopathy(EAST)syndrome]罕见,病因为Kir4.1/Kir5.1突变。患者有Gitelman样表型、失盐和低钾性碱中毒表现,故应与Bartter综合征/ Gitelman综合征鉴别。
家族性低镁血症伴高尿钙-肾钙质沉着症
家族性低镁血症伴高尿钙-肾钙质沉着症(familial hypomagnesaemia with hypercalciuria and nephrocalcinosis,FHHNC)为常染色体隐性遗传,病因与paracellin-1(PCLN-1)突变相关,主要表型有低镁血症、高尿钙、肾钙质沉着、低渗尿和进行性肾衰竭。
根据血压和酸碱平衡状况鉴别遗传性低钾血症的病因
慢性低钾血症的鉴别方法很多,此处介绍的鉴别方法可以从临床表现一直追溯到病因,尤其是对遗传性低钾血症的鉴别很有帮助。一般可以分为以下4步进行(图4-20-2):
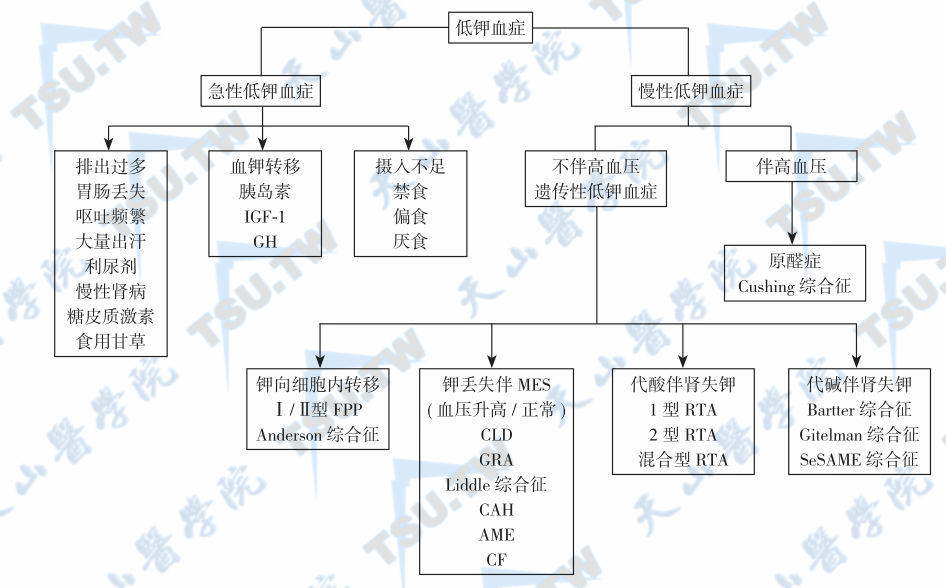
遗传性低钾血症的病因与鉴别
注:GRA: glucocorticoid-remediable aldosteronism,糖皮质激素可治疗性醛固酮增多症; FPP:familial periodic paralysis,家族性周期性麻痹;CLD:congenital chloride-losing diarrhea,先天性失氯性腹泻; CF: cystic fibrosis,囊性纤维化;CAH:congenital adrenal hyperplasia,先天性肾上腺皮质增生症;AME:apparent mineralocorticoid excess,表观盐皮质激素过多;MES:mineralocorticoid excess state,盐皮质激素过多状态; SeSAME:seizures-sensorineural deafness-ataxia-mental retardation and electrolyte imbalance,惊厥-感觉神经性耳聋-共济失调-智力障碍和电解质紊乱综合征
- 第一步:根据血压情况,首先分为低钾血症伴高血压和低钾血症不伴高血压两类;
- 第二步:低钾血症伴高血压的病因鉴别可搜索相关篇幅;
- 第三步:低钾血症不伴高血压的病因查找是临床上的难题,有时相当困难;因此需要同时根据血气分析结果(分为不伴酸碱平衡紊乱、伴有代谢性碱中毒和伴有代谢性酸中毒3种)将遗传性低钾血症分为4类,即钾向细胞内转移(家族性低钾血症性周期性瘫痪)、肠或肾钾丢失伴盐皮质激素过多状态、代谢性碱中毒伴肾小管失钾和代谢性酸中毒伴肾小管失钾(下表)。肠或肾钾丢失伴盐皮质激素过多状态患者如果已经有血压升高,则进入低钾血症伴高血压的鉴别程序;
- 结合其他相关病史和临床特征,限定可能的遗传病因,并用致病基因突变分析最后确定诊断。
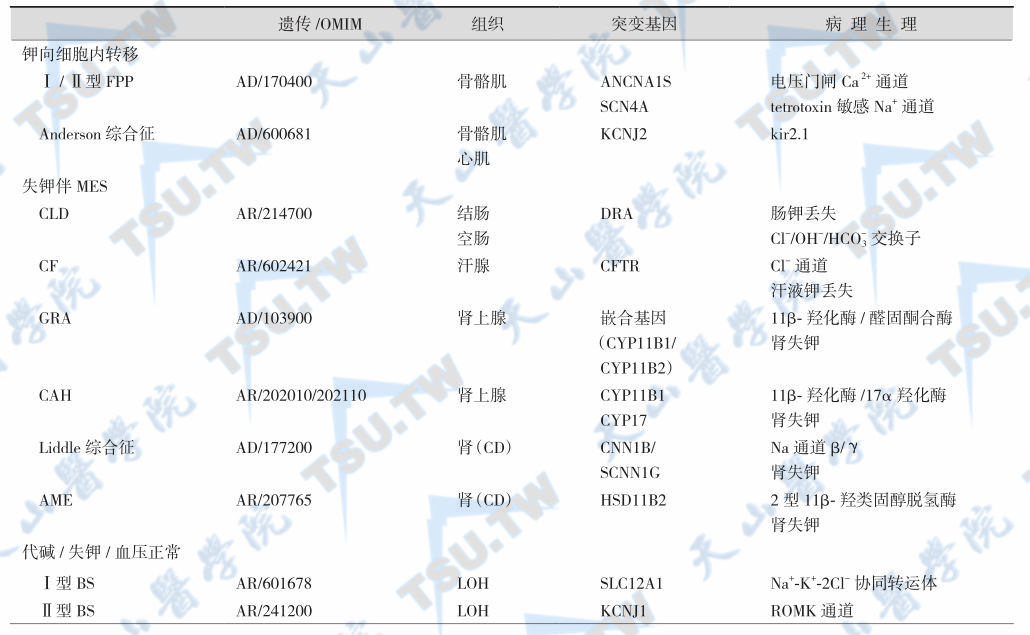
遗传性低钾血症的病因与鉴别
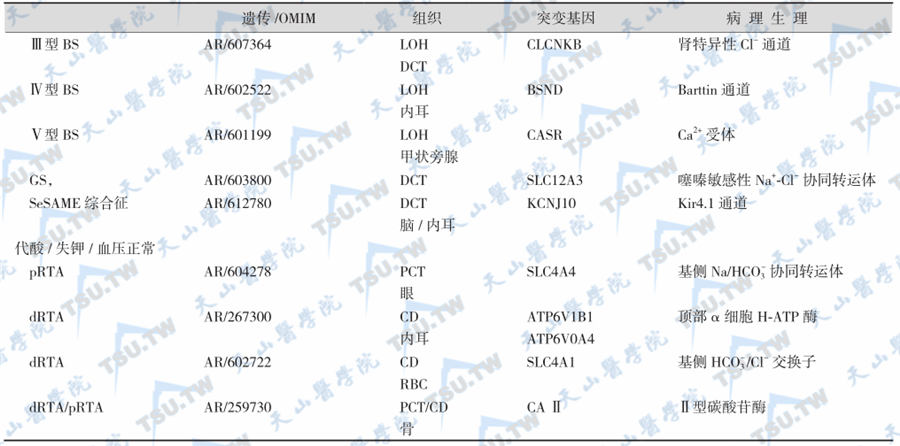
续表
- PC:proximal convoluted tubule,近曲小管;
- LOH:loop of Henle,Henle袢;
- DCT:distal convoluted tubule,远曲小管;
- CD:collecting duct,集合管;
- AD:autosomal dominant,常染色体显性遗传;
- AR:autosomal recessive,常染色体隐性遗传;
- RBC:red blood cell,红细胞;
- GRA:glucocorticoid-remediable aldosteronism,糖皮质激素可治疗性醛固酮增多症;
- FPP:familial periodic paralysis,家族性周期性瘫痪;
- CLD:congenital chloride-losing diarrhea,先天性失氯性腹泻;
- CF:cystic fibrosis,囊性纤维化;
- CAH:congenital adrenal hyperplasia,先天性肾上腺皮质增生症;
- AME:apparent mineralocorticoid excess,表观盐皮质激素过多;
- BS:Bartter syndrome,Bartter综合征;
- GS:Gitelman syndrome,Gitelman综合征;
- pRTA:proximal renal tubular acidosis,近曲小管性酸中毒;
- dRTA:distal renal tubular acidosis,远曲小管性酸中毒;
- MES:mineralocorticoid excess state,盐皮质激素过多状态;
- SeSAME:seizures-sensorineural deafness-ataxia-mental retardation and electrolyte imbalance,惊厥-感觉神经性耳聋-共济失调-智力障碍和电解质紊乱综合征
