
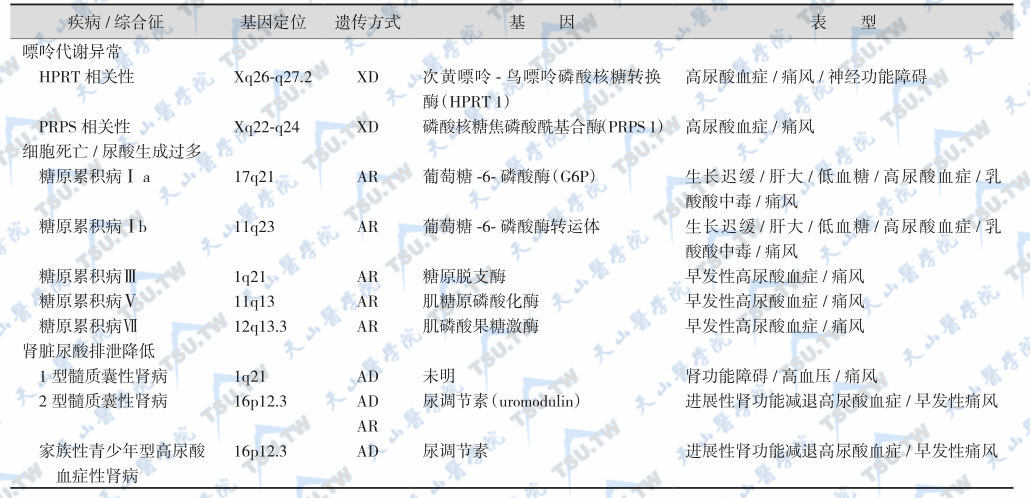
大多数原发性高尿酸血症的病因未明,少数由于酶缺陷引起,发病与尿酸生成过多或肾尿酸排泄减少有关。在有痛风史的家庭中,无症状高尿酸血症者占25%~27%。Hange和Harvald研究了32例痛风患者的261个亲属,6.1%有痛风。另两项关于痛风家族的研究显示,25%有高尿酸血症,而正常人群仅4.6%。高尿酸血症的分布方式与多基因遗传有关,O’Brian等对Blackfeet和Pima Indians的研究显示,血尿酸水平由多基因决定,伴高尿酸血症/痛风的孟德尔遗传综合征病因和临床表现见下表。
伴有高尿酸血症/痛风的孟德尔遗传综合征
人尿酸转运子-1(URAT1)是尿酸-阴离子的交换子,可对血尿酸水平进行调节,编码基因为SLC22A12(定位于11q13),其失活性突变导致特发性低尿酸血症。遗传性低尿酸血症(hereditary hypouricemia)的一个重要原因是URAT1突变,使尿酸重吸收障碍,尿液中尿酸升高而血尿酸降低,常伴有尿酸结石、肾损伤甚至肾衰竭。
高尿酸血症具有家族聚集和地域流行特征
太平洋地区,尤其是我国台湾省的土著居民的发病率较高。高尿酸血症和痛风是常染色体多基因显性遗传病,可能存在导致疾病发生的易感基因或者致病基因。在4号染色体4q25区存在高尿酸血症或痛风的易感基因。家族性肾病伴高尿酸血症(familial nephropathy associated with hyperuricemia)为常染色体显性遗传性疾病,其病因与UMOD基因突变有关,多见于西班牙,主要表现是高尿酸血症、痛风、肾功能不全和高血压,但表现很不均一。肾损害以间质性肾病为特点。尿调节素(uromodulin)相关性肾病(uromodulin-associated kidney diseases)表现为家族性青少年高尿酸血症性肾病和囊性髓质肾病,常染色体显性遗传。患者有高尿酸血症、痛风和进行性肾功能不全,尿中尿调节素的排泄减少。
全球的高尿酸血症和痛风发病率呈增加趋势。1 项2046例健康男性15年的调查发现,血清尿酸水平在536.5μmol/L(9mg/dl)以上者,年痛风发病率为4.9%;而尿酸值在420~530μmol/L之间者发病率降至0.5%,血尿酸在420μmol/L以下者发病率0.1%。Framingham研究中,血尿酸420~470μmol/L者仅12%发展为痛风;而大于535μmol/L者,痛风发病率增加6倍,但这些患者仅代表痛风人群的20%。不同种族人群之间患高尿酸血症与痛风的易患性差异较大。随着饮食结构改变及人均寿命延长,高尿酸血症和痛风的患病率逐渐升高。1996~1997年我国上海黄埔地区流行病学调查显示,该地区高尿酸血症患病率为10.1%,男性14.2%;痛风患病率0.34%,男性0.77%,与1980年调查的结果比较大幅度上升。
嘌呤代谢障碍导致尿酸生成增多
限制嘌呤饮食5天后,如尿酸排泄量>600mg/d,可认为是尿酸生成过多。尿酸生成增多的主要原因是嘌呤代谢酶缺陷。
次黄嘌呤-鸟嘌呤磷酸核糖转移酶活性降低
次黄嘌呤-鸟嘌呤磷酸核糖转移酶(HGPRT)由HPRT基因编码,HPRT基因位于X染色体q26-27,长57bp。HGPRT是嘌呤补救合成途径的关键酶,HPRT突变导致HGPRT活性降低,使鸟嘌呤转变为鸟嘌呤核苷酸和次黄嘌呤核苷酸减少,两种嘌呤不能合成核苷酸或被清除而使血尿酸升高。目前已发现外显子1~9存在2000余种突变位点,外显子3、8是突变的“热点区”。根据突变致酶活性降低的程度可分为HGPRT严重缺陷(Lesch-Nyhan综合征。hUAT至少有2个跨膜区,并形成两个细胞外区,各含1个β-半乳糖苷结合位点。第1、2跨膜螺旋间有2个β片层,第2、3跨膜α-螺旋则形成发夹样结构,为尿酸盐的结合位点。hUAT蛋白存在多个特殊功能区域,不同物质与不同位点结合后都可影响hUAT功能。hUAT的两个β-半乳糖苷结合位点位于胞外侧,与特异性底物结合后能改变hUAT蛋白功能。D(+)-葡萄糖是人体内的生理性葡萄糖,虽然对β-半乳糖苷结合位点的亲和性比α-乳糖低,但所起的作用与α-乳糖相同,提示D(+)-葡萄糖调节hUAT通道活性。尿酸酶的特异阻断剂oxonate、抗结核药物吡嗪酰胺(pyrazinamide,PZA)或腺苷可阻断hUAT通道活性。研究表明,hUAT可能是与尿酸盐分泌密切相关的转运蛋白,其基因突变和(或)多态导致hUAT功能障碍,引起高尿酸血症和痛风。
hURAT1
hURAT1含10个内含子和9个外显子,cDNA全长2642bp,编码区1659bp,编码含555个氨基酸残基的蛋白质。hURAT1是有机阴离子转运子家族(OATs)的类似物,其氨基酸序列与OAT4同源。hURAT1蛋白主要位于肾皮质近曲小管的上皮细胞,具有转运尿酸盐功能。许多有机阴离子,如乙酰乙酸、琥珀酸盐、吡嗪酰胺(PZA)等都可影响尿酸盐经hURAT1的转运过程。G774A突变者血尿酸显著降低,Cua/Ccr显著升高。在正常人,促尿酸排泄药物PZA、苯溴马隆、丙磺舒均反式促进尿酸盐经hURAT1的摄取,说明hURAT1是促尿酸排泄药物的作用靶点,与大多数遗传性肾性低尿酸血症的发生有关。
有机阴离子转运子1
有机阴离子转运子1(OAT1)编码含551个氨基酸残基的蛋白质,有12个跨膜区,主要表达于肾脏。OAT1为对氨基马尿酸(para-aminohippuric acid,PAH)、α-酮戊二酸、促尿酸和抗尿酸排泄药物(如丙磺舒、苯溴马隆)等的作用靶点。OAT1基因突变与家族性青年性痛风性肾病有关。
hOAT3
hOAT3与OATs有相同结构,属于OATs家族成员,表达于肝脏、肾脏、脑及眼组织,有12个跨膜区,4个N-糖基化位点,8个依赖蛋白激酶C的磷酸化位点。OAT3具有跨膜转运功能,是有机阴离子/二羧酸盐交换子,参与肾脏尿酸盐转运。
尿酸具有强烈的抗氧化与促氧化作用
以前,人们认为尿酸是一种代谢废物,当形成结晶后,可以诱发肾石病和痛风性关节炎。继而认识到,尿酸是一种强氧化剂和NOS辅酶,可以清除单线态氧(singlet oxygen)、氧自由基(oxygen radicals)、过氧化亚硝酸盐(peroxynitrite),螯合过渡金属物(chelates transition metals),降低铁离子介导的抗坏血酸氧化(iron ion-mediated ascorbic acid oxidation),防止脂质和蛋白质过氧化,灭活四氢生物蝶呤,尿酸盐约占血浆总抗氧化能力的50%,在抗心血管病、衰老和肿瘤中起了重要作用(下图)。
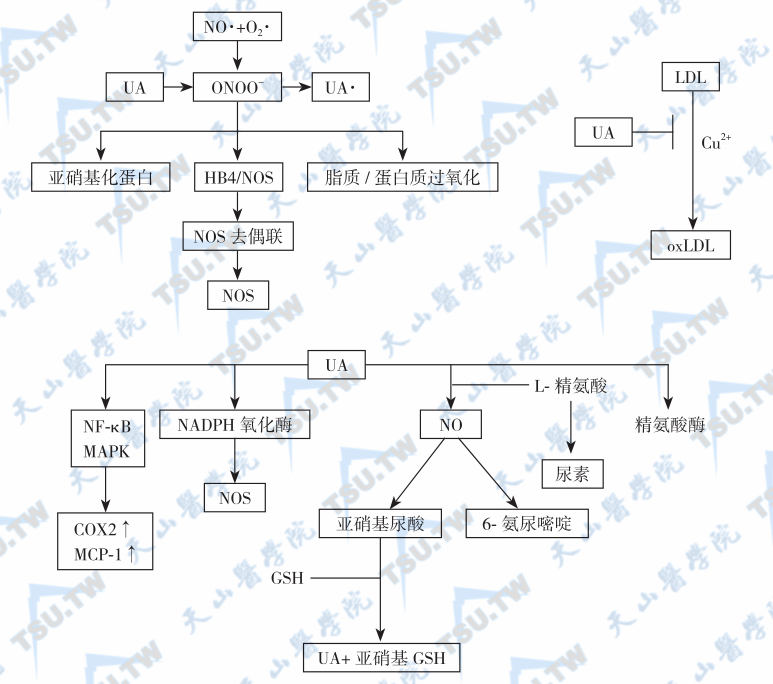
尿酸的抗氧化作用
注:左上图表示尿酸的抗氧化作用,右上图表示尿酸抵抗铜离子引起的脂质氧化,下图表示尿酸增强精氨酸酶活性,并能使精氨酸生成NO途径转向生成尿素途径。ONOO-:Peroxynitrites,过氧化亚硝酸盐;NO·:nitric oxide,一氧化氮;O2·:superoxide,超氧化物;HB4:tetrahydrobiopterin,四氢生物蝶呤;UA :Uric acid,尿酸;UA·:uric acid radicals,尿酸根;oxLDL:oxidation of LDL,氧化型LDL;GSH:glutathione,谷胱甘肽。
但是在特定的化学微环境(chemical microenvironment)中,尿酸既是促氧化剂(pro-oxidant),也是抗氧化剂(antioxidant)。体外环境和离体细胞研究发现,尿酸增加LDL 氧化和氧化型LDL颗粒再氧化。当尿酸被氧化时,产生的尿酸酸根可扩增促氧化效应,但这一作用可被抗坏血酸中和。NO降低诱发高血压和胰岛素抵抗,在抗坏血酸作用下,尿酸盐可与NO直接反应而生成不稳定的亚硝基尿酸(nitrosated uric acid),最后生成稳定的6-氨基尿嘧啶(6-aminouracil)。因此高尿酸血症时,可降低NO的生物可用性,引起尿酸微结晶和组织炎症,破坏关节软骨、肾组织和血管内皮,导致一系列病变。
