
雄激素受体
雄激素对正常雄性发育、分化及功能维持十分必要。雄激素的作用机制与其他类固醇激素相似,即雄激素与胞质中的受体结合形成雄激素-受体复合物,通过雄激素受体(androgen receptor,AR)的介导而起作用。AR是类固醇受体超家族中的典型成员,能以高专一性、高亲和力与激素应答元件结合调控基因转录。AR是一种核蛋白,由918个氨基酸组成,相对分子质量约为99 000,沉降系数为8.3S。编码AR的基因于1988年由Lubahn和Chang等首次克隆成功,并定位于X染色体的长臂上(Xq1112),长7590kb,由8个外显子组成。外显子1最大,编码受体的N端,N端的结构最不保守。结构分析表明,N端结构域与AR的转录激活有关。这个区域包含两种多聚体——多聚谷氨酸和多聚脯氨酸,多聚氨基酸结构被认为在转录激活方面起重要作用。外显子2和3编码AR的DNA结合结构域(DBD),该结构域高度保守,由68个氨基酸组成,能折叠成两个锌指(zinc finger)结构。外显子4~8编码受体的铰链区和配体结合结构域(LBD),该区域起着形成二聚体和结合配体的作用。
AR发挥其生物学功能时首先要与其靶基因上的雄激素应答元件(ARE)相互作用。目前对ARE的研究相对较落后,已发现的ARE基本与糖皮质激素受体应答元件一致。迄今为止,已有多个甾体激素的辅助激活因子被克隆,但AR专一的辅助激活因子只有一个,而且其作用机制尚不十分清楚。
雄激素受体异常与疾病
目前已知与AR突变相关的疾病有雄激素不敏感综合征、延髓脊髓性肌萎缩、前列腺癌和男性乳腺癌等,随着对AR的深入研究,这些疾病的分子基础日益明了。这包括完全的或部分的基因缺失、点突变、mRNA剪接位点突变,插入突变以及谷氨酸多聚结构的增大等。在此主要介绍雄激素不敏感综合征。
雄激素不敏感综合征(androgen insensitivity syndrome,AIS),也称为雄激素抵抗征。这是由于AR缺陷致使靶细胞对雄激素反应能力低下或无反应的一类X连锁隐性遗传病。患者染色体核型为46,XY,H-Y抗原呈阳性。
发病机制
AIS系AR编码基因的部分缺失、点突变或受体的mRNA转录过程受损,导致AR功能的不同损害,因此使男性表型分化和/或男性化异常。自1988年第一例AIS患者AR基因突变被报道以来,迄今为止已有110多种AR基因突变被报道。这些突变发生在AR基因的8个外显子上都有发现,但大多数突变发生在AR的LBD及DBD。在LBD有两个错义突变高发区(分别位于726-772及826-846氨基酸残基之间)。目前已确定的AR基因突变按所造成的AR表型可分为下列4类:
- 受体阴性型:其分子机制为:a.AR基因全部或部分丢失,较少见,分别占总突变的1%和4%;b.无义突变造成AR合成提前终止,由此生成的截短受体因C段的部分缺失,无激素结合能力;c.内含子与外显子边界区的突变造成RNA拼接位点改变,致使基因转录后的mRNA加工发生错误;d.错义突变,如LBD区的某些氨基酸的改变,造成受体无激素结合能力,这说明某些氨基酸对维持LBD的功能非常重要;
- 受体结合减少型:这主要是由于AR数量的减少或雄激素对AR亲和力降低所致。如Zoppi报道的Gln的无义突变,所合成的受体是以Met为翻译起点,而这一合成效率大大下降,仅为正常的25%左右,且变异受体转录激活能力也下降;
- 受体热不稳定型:Marcelli报道的一个突变(Pro→Ser)就为此机制,突变受体热稳定性明显下降且与配体的解离率增加;
- 受体正常型:这种类型的突变大都发生在受体的DBD区。例如Arg→His等,因Arg 在DBD中高度保守,这种突变会造成AR的功能丧失。
临床表现
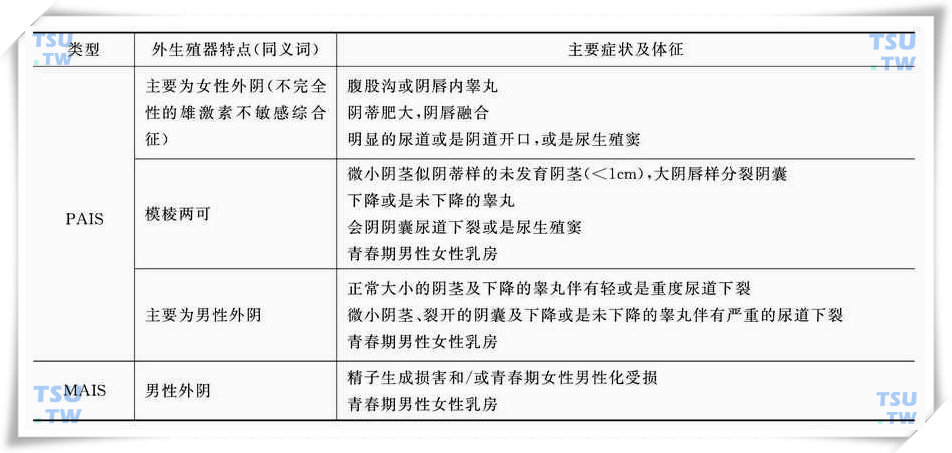
雄激素不敏感综合征表型的变化很大,自常见的睾丸女性化至单纯尿道下裂。雄激素不敏感综合征典型的症状是出生时外生殖器女性化,青春期第二性征发育异常,患者核型为46,XY,不育。雄激素不敏感综合征被分为3型:完全性雄激素不敏感综合征(complete androgen insensitivity syndrome,CAIS),典型的女性外阴;部分性雄激素不敏感综合征(partial androgen insensitivity syndrome,PAIS),外阴主要表现为女性外阴或是男性外阴或是外阴模棱两可;轻度雄激素不敏感综合征(mild androgen insensitivity syndrome,MAIS),典型的男性外阴。分型及临床表现见下表。
雄激素不敏感综合征分型及临床表现

临床诊断
根据临床表现及患者的染色体核型46,XY,可以获得初步诊断。实验室检查患者的睾酮水平正常或是升高,并能正常的转化为双氢睾酮,LH正常或是升高,外生殖器皮肤纤维母细胞雄激素结合活性缺乏。分子遗传学方面的主要是检查AR基因,超过95%的CAIS的患者AR基因发生了突变,不到50% 的PAIS的患者AR基因发生了突变。CAIS患者在出生后的3个月之内血清中的LH及T的峰值降低。
临床治疗
治疗CAIS时,为了防止睾丸的恶变,要么待女性化完成后切除睾丸,要么青春期前行性腺切除术并进行雌激素替代治疗,同时应进行阴道扩张术,避免以后的性交困难。PAIS外阴女性化显著的患者治疗方案同CAIS,PAIS外阴女性化显著的患者最好是在青春期以前进行性腺切除术,避免青春期时阴蒂肥大。对于PAIS外阴模棱两可或是男性化显著的患者应该尽早确定按哪种性别进行抚养,确定当作男性抚养的患者应该行泌尿系手术,如睾丸下降固定术及尿道下裂修补术;确定当作女性抚养的患者应该行青春期后性腺切除,并给予雌激素及雄激素替代治疗。MAIS患者需要行乳房整形术。婴幼儿时期补充睾酮治疗可以帮助改善女性男性化。
