
雄激素不敏感综合征(androgen insensitivity syndrome,AIS)属于46,XY性发育障碍(DSD),是一组与雄激素受体(androgen receptor,AR)缺陷有关的遗传性性发育障碍综合征的总称,属于DSD的范畴。雄激素在靶细胞作用过程的任何一个步骤发生异常都可引起雄激素的作用不完全和男性假两性畸形。临床上,主要包括雄激素受体缺陷症(androgen receptor deficiency)、5α-还原酶缺陷症(5α-reductase deficiency)、芳香化酶缺陷症(aromatase deficiency)和雌激素受体缺陷症(estrogen receptor deficiency)4种。雄激素受体缺陷症的病因和发病机制如下:
雄激素受体功能障碍分为五种类型
雄激素受体(AR)基因突变引起的AR功能障碍有下列5种类型。
- 雄激素与AR结合障碍:突变主要发生于4、5、7、8号外显子,其中以第7和第8号外显子的突变报道最多,多数患者表现为完全性睾丸女性化,少数患者表现为不完全性睾丸女性化,极少数仅表现为尿道下裂。
- 雄激素-雄激素受体复合物与DNA结合障碍:突变主要发生于第2和第3号外显子,多数患者表现为完全性睾丸女性化,部分患者表现为不完全性睾丸女性化或伴有前列腺癌。
- 受体蛋白分子截短:突变位于内含子/外显子接合点,这类突变发生率低(如第8号外显子的突变,AGC→AGT),使转录的DNA截短5kb,表达的AR蛋白链缩短,而且还伴有突变点后的若干个氨基酸的错义替代,患者多表现为不完全性睾丸女性化。
- 配体特异性改变:这类突变(如M807T和T877A)少见,但不一定发生在激素结合区,突变改变了AR与雄激素的结合性能,突变型AR与睾酮或二氢睾酮的结合亲和力差,但与孕酮及其他类固醇激素的结合亲和力反而很高。还有一类突变(如N223K)可使AR对热不稳定,临床上可表现为男性不育症或部分性AIS。
- AR后信号转导缺陷:在正常情况下,AR的N端和C端存在相互作用,并对AR后的信号转导有调节作用。一些突变(如M742V、F725L、G743V、F754L和M886V等)使AR的N端与C端的相互作用异常,使患者仅有轻度AR功能失常的实验室表现,不引起完全型AIS,在大剂量雄激素的诱导下,可使AR功能有所恢复。
雄激素受体突变引起46,XY-DSD
雄激素受体的结构件见下图。因为雄激素受体定位于X染色体,因而AR突变为X-性连锁隐性遗传。已报道的AR基因突变位超过600个(http://www.mcgill.ca/ androgendb),涉及AR基因所有外显子,但大多数突变在DNA结合区和配体结合区;突变类型包括序列插入、提前终止编码、mRNA剪接异常、单个碱基替代、缺失和框架移动等,其中外显子2、3、7和8的点突变最为常见,引起46,XYDSD(睾丸女性化)。具有46,XY染色体核型的男性胎儿在胚胎性发育中缺乏雄激素的作用,使中肾管及泌尿生殖窦不能分化为男性生殖管道而发育为女性表型。
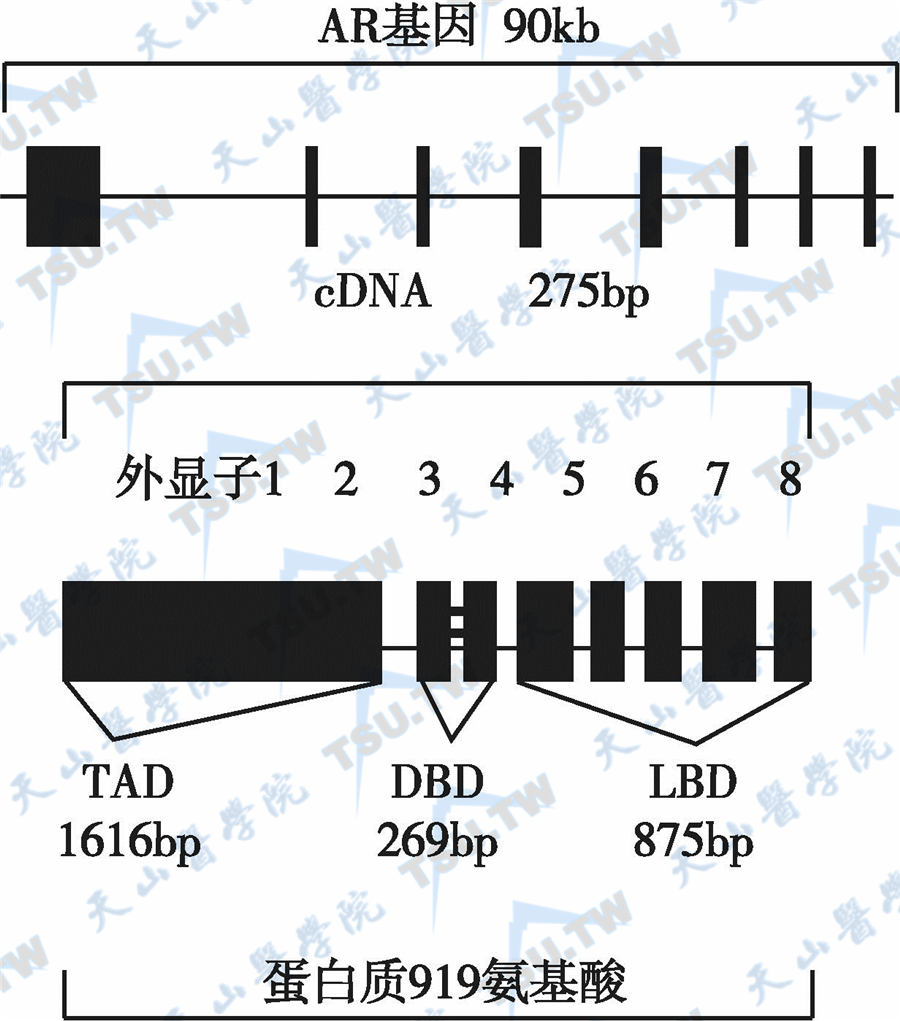
AR基因结构的示意图
系统的医学参考与学习网站:天山医学院, 引用注明出处:https://www.tsu.tw/edu/3900.html
