
症状表现组成连续表型谱
本病有家族发病倾向,但1/3患者无家族史。临床表现不均一。根据AIS的程度,46,XY-DSD可分为完全型AIS(完全性睾丸女性化,女性表型)、部分型AIS(部分性睾丸女性化,外生殖器两性畸形)和混合型AIS(不育男性)3类。但是,AIS的雄激素敏感程度是一个连续的表型谱(phenotype spectrum),部分性AIS还可以分出许多亚类,见下图。但是,为了诊断方便,一般将AIS分为完全型睾丸女性化、不完全型睾丸女性化和男性外生殖器伴不育3型。

雄激素敏感程度分级
完全型睾丸女性化
纯合子(完全型睾丸女性化,complete testicular feminization)患者的典型表现是染色体性别与外生殖器性别和(或)社会性别完全相反。睾丸位于阴唇阴囊中或腹股沟处。男性婴儿具有阴蒂样小阴茎,会阴型或阴囊型尿道下裂。睾丸位于阴唇阴囊中或腹股沟处,无子宫和输卵管等。前列腺缺如或发育不良。附睾、输精管和精囊腺等分化良好。在出生时,往往被当作女性抚养。
部分型睾丸女性化
约1/3的部分型睾丸女性化(partial testicular feminization)患者出生时为假的女性外阴(阴道尿道下裂与假阴道);约1/3为尿生殖窦存留,55%伴盲管阴道,但也可仅表现为单纯性尿道下裂或盲管阴道。不完全性睾丸女性化是AIS的另一种类型,由于AR的不完全性缺陷,临床表现为外生殖器两性畸形,畸形的程度可因具体患者而异。典型病例出生时为男性外阴,但存在小阴茎、阴囊型或会阴型尿道下裂及隐睾等严重的男性化不全表现。附睾和输精管发育不全或正常,有盲管阴道,无子宫和输卵管。青春期阴毛、腋毛生长,男性乳腺发育,睾丸小,无精子生成。
较轻的患者可仅表现为不育或小阴茎、分叉阴囊和尿道下裂;较重者有严重尿道下裂和盲管阴道。如果在会阴部有依赖于雄激素的终毛存在,则为不完全性睾丸女性化,但量少于正常人。
男性外生殖器伴不育
男性外生殖器伴不育(male external genitalia with infertility)患者的性功能和性行为及外生殖器正常,但精子生成和精子功能异常,缺乏生育能力。因为没有第二性征异常,故很难做出早期诊断,仅在婚后检查不育原因时才被发现存在AR基因突变,而雄激素的敏感性有轻度降低。
46,XY-DSD以雄激素不敏感为特征
患者的外生殖器与体型开始男性化,可具相当程度的男性体型,骨骼粗壮,肌肉较发达,皮下脂肪少,出现喉结、嗓音低沉、阴茎增大,可勃起和射精。阴唇阴囊的皮肤出现粗的皱褶,且有色素沉着。但男性化发育不全,阴毛、腋毛和胡须稀少,两鬓发际不退缩、痤疮少见。除个别患者外,一般无男性乳腺增生,这是因为本病患者睾酮和E2的产生率均正常,没有发生两者比例失调的结果。患者有射精,但精液量减少。染色体核型46,XY,外生殖器为女性型,盲管阴道,无子宫和输卵管,睾丸位于腹腔、腹股沟管或阴唇内,无附睾和输精管。青春期出现乳腺发育,大小和正常女性相似,外阴也发育,但呈幼稚型,小阴唇发育差,阴蒂正常或稍小,无腋毛和阴毛生长或极稀少,无月经来潮。
本病患者无生育能力。雄激素替代治疗可维持男性第二性征。异位睾丸,特别是位于腹腔内者易发生癌变,包括精细胞癌、性腺母细胞瘤、恶性胚胎瘤和恶性Leydig细胞肿瘤等。故本病如长期未被确诊及治疗,睾丸可恶变。骨密度降低,但可达中等最终身高。
从男性外生殖器异常病例中筛查46,XY-DSD
凡遇下列情况之一均应考虑46,XY-DSD可能:①新生儿外阴男女性别难辨或无睾丸;②青春期无月经来潮而外生殖器接近女性,阴毛和腋毛缺如或稀少,盲管阴道和无子宫等而染色体核型46,XY;③睾丸滞留于腹股沟,伴或不伴发腹股沟疝,或睾丸在“大阴唇”或阴唇阴囊内;④外阴接近于正常男性,青春期有乳腺发育;⑤患特发性男性不育症或原因不明的男性乳腺发育;⑥血浆LH和睾酮增高(但在婴儿期可以正常);⑦性功能减退。
患者就诊的主要原因是青春期缺乏乳腺发育和月经。完全性睾丸女性化主要应与部分性雄激素不敏感综合征(partial androgen insensitivity syndrome,PAIS)或3型17β-羟类固醇脱氢酶(17β-hydroxysteroid dehydrogenase type 3,17β-HSD3缺陷症鉴别。外生殖器两性畸形应与其他原因引起者鉴别。应对“腹股沟疝”女婴进行详细检查,因为1%~2%的完全型46,XY-DSD(完全型AIS)以“疝”为突出表现。在青春期,对原发性闭经女性体查时,要特别注意乳腺发育和阴毛分布状态,并详细检查外生殖器,排除Mayer-Rokitanskyküster-Hauser综合征(苗勒管发育不全症),如仍未能诊断,则行染色体核型分析(karyotype analysis),并用MRI或盆腔B超对内生殖器进行探查。完全性完全型46,XY-DSD应与Swyer综合征、XY完全型性腺发育不良鉴别,缺乏乳腺发育和身材短小为其特点。青春期完全性完全型46,XY-DSD患者的血睾酮和LH升高,常伴雌二醇升高(MRKH综合征患者正常),此时必须行染色体核型分析或雄激素受体基因分析。
完全性睾丸女性化、Reifenstein综合征和SRD5A2缺陷症均有外生殖器两性畸形,其鉴别要点是激素分泌特点与分子病因,见下表。
三种雄激素抵抗综合征的鉴别
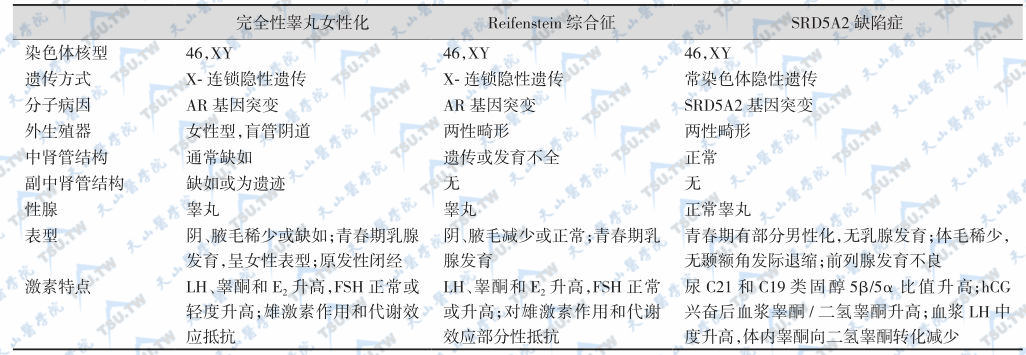
注:AR:雄激素受体;SRD5A2:2型5α-还原酶;LH:黄体生成素;FSH:促卵泡激素;E2:雌二醇
激素测定和垂体-性腺轴动态试验提示诊断
FSH和LH
血清FSH大多正常或轻度增高,但LH升高明显。注射GnRH后LH有过度反应,LH和睾酮显著升高是AR基因突变致46,XY-DSD的诊断依据之一。AR基因突变致睾丸女性化的诊断依据是:①女性体型,青春期乳腺发育良好,无阴毛和腋毛,无月经来潮,外阴发育不良;②盲管阴道,无子宫;③在大阴唇或腹股沟管内可触及睾丸;④LH和睾酮显著升高;⑤染色体核型46,XY,青春期前可作HCG兴奋试验,有助于本病的早期诊断;⑥确诊有赖于AR数目和功能测定,病因诊断可作AR基因突变检测。
血清睾酮和去氢异雄酮硫酸盐
血清睾酮和去氢异雄酮硫酸盐(dehydroepiandrosterone sulfate,DHEAS)可以分别认为是卵巢性与肾上腺性高雄激素血症的标志物,而二氢睾酮对诊断和鉴别诊断没有帮助,因为皮肤的皮脂腺含有丰富的5-α还原酶,可将睾酮转化为二氢睾酮和雄烯二酮。但是,二氢睾酮的终末代谢产物却是诊断高雄激素血症和多毛的良好标志物。
HCG兴奋试验
适用于怀疑有17β-羟类固醇脱氢酶(17β-hydroxysteroid dehydrogenase,17β-HSD)缺陷患者与雄性化不足患者的诊断。方法:在HCG兴奋前后分别测定血清睾酮(testosterone,T)与雄烯二酮(androstenedione,A)比值(T/A比值),必要时延长HCG兴奋时间。据Faisal等报道,完全性AIS患者,基础和HCG兴奋后T/A比值分别为0.4 (0.1~8.0)和4.5(0.5~16.7);部分性AIS分别为0.7(0.1~15.0)和3.9(0.3~20.5);睾丸发育不全或病变者分别为0.4(0.1~5.6)和0.6(0.1~3.6),兴奋后的T/A比值明显低于正常人。绝大多数AR突变所致AIS的T/A比值明显升高,个别(4/84)<0.8,但延长HCG兴奋时间后,T/A比值可升高。因此,在46,XY男性中,如果HCG兴奋后的T/A比值<0.8,应考虑17β-HSD3缺陷症(17β-hydroxysteroid dehydrogenase 3 deficiency)可能(睾丸组织功能异常除外)。这一试验可用于AR或17β-HSD3基因突变分析前的病例筛选(应先排除睾丸病变)。
HCG可刺激睾酮分泌,睾酮升高则可抑制性激素结合球蛋白的合成,使血液循环中这种蛋白降低。HCG-睾酮联合试验可用于评估睾酮的敏感性。该试验的原理是以注射睾酮后血中性激素结合球蛋白降低的百分率来评估细胞对睾酮的敏感性,方法是平衡试验。试验的第1天和第4天各肌注HCG 2500U,同时在这之前采血测血清性激素结合球蛋白,于试验的第7天采血测血清睾酮,继而肌注庚酸睾酮(testosterone enanthate),2mg/kg,于第14天再采血测血清性激素结合球蛋白,并与注射HCG之前所测基础值比较,计算出注射睾酮后性激素结合球蛋白下降的百分率。如下降率大于80%则为试验阳性。此试验对诊断青春期前而AR阳性患者有价值。AIS因细胞对睾酮不敏感,故注射睾酮后,性激素结合球蛋白不下降,故试验为阴性。注射HCG的目的在于排除睾酮合成缺陷所致疾病。
根据雄激素受体数目/功能/突变分析确立诊断
一般采取外阴皮肤成纤维细胞进行体外培养,然后加入用氚标记的睾酮或二氢睾酮,测定受体结合容量和亲和力,以了解AR在量和质方面的改变;或取患者的外生殖器皮肤成纤维细胞培养,检查AR与雄激素结合情况。根据有无结合的结果可分为AR结合阳性和AR结合阴性两类。
受体结合容量和亲和力测定
完全性睾丸女性化患者大多数为结合阴性,常见于AR配基结合区突变;单纯性男性不育症、尿道下裂或单纯性男性乳腺发育者AR结合多为阳性,常由于DNA结合区突变所致;部分性睾丸女性化者的AR结合试验结果可为阳性,亦可为阴性;即使表型为完全性睾丸女性化的患者,结合试验亦可为阳性。少数睾丸女性化患者雄激素与其AR的结合量和性质均无改变,这些患者可能存在受体后缺陷或对雄激素作用有重要影响的其他基因突变。
雄激素受体功能测定
AR的功能改变包括:①AR亲和力减低,表现在结合后易于离解,可测定离解常数(kd);②雄激素与AR结合后的复合物对热不稳定,反映在温度升高到42℃时,结合量则下降到37℃时所测结合量的20%以下;③用整体细胞或细胞核与用3H-标记的睾酮或二氢睾酮温育,后者与核中特异性DNA结合量减少或缺如;④AR复合物不能外形变构而引起静电荷改变,使之不能与阴离子多的DNA结合;⑤AR与雄激素结合力下降,但与孕激素呈高亲和力结合;⑥分子筛色谱层板及ZD凝胶电泳图异常;⑦继发性SRD5A2活性下降。
AR基因检测
能证明患者的AR基因突变。
雄激素作用障碍与2型5α-还原酶缺陷/CYP19芳香化酶缺陷/雌激素受体缺陷症/无睾症鉴别
雄激素作用障碍的病因主要有4种:①雄激素受体基因突变;②2型5α-还原酶缺陷症(2型5α-还原酶基因突变);③CYP19芳香化酶缺陷症(CYP19芳香化酶基因突变);④雌激素受体缺陷症。因此,雄激素受体基因突变引起的外生殖器两性畸形与雄激素缺乏综合征必须与2型5α-还原酶缺陷症和CYP19芳香化酶缺陷症鉴别。
2型5α-还原酶缺陷症
SRD5A2缺陷症于1961年首先由Nowakowski和Lenz报告,是一种常染色体隐性遗传疾病。人类有2种类固醇5α-还原酶(SRD5A),即SRD5A1 和SRD5A2。两者的氨基酸序列约50%相同,以NADPH为辅因子,催化睾酮转化为作用更强的二氢睾酮。SRD5A1由295个氨基酸残基组成,基因位于5p15,含5个外显子,在肝脏和非生殖器皮肤表达。SRD5A2由254个氨基酸残基组成,基因位于2p23,亦有5个外显子,主要在外生殖器皮肤和前列腺表达。睾酮通过弥散作用进入靶组织细胞,在胞质液中大部分睾酮被SRD5A转化为二氢睾酮,二氢睾酮与AR结合后进入细胞核,再与DNA受体结合,激活基因转录,合成特异性蛋白。在胚胎期,发育成男性外生殖器的生殖结节、生殖膨隆和泌尿生殖窦的胚胎细胞中有SRD5A2,而在中肾管细胞中则缺如,因此,决定男性外生殖器分化发育的雄激素主要是二氢睾酮。SRD5A2缺陷使睾酮不能转变为二氢睾酮,二氢睾酮缺乏使尿生殖窦和前列腺等依赖二氢睾酮的器官和组织发育发生障碍,致外生殖器不能发育成男性而呈两性畸形。
SRD5A2缺陷症是SRD5A2基因突变所致,有3种类型:①酶无活性;②酶不稳定,迅速被代谢;③酶活性降低和降解加速兼有。SRD5A2基因突变的类型很多,主要包括单碱基突变、单碱基插入和碱基缺失等。大多数患者为纯合子突变,少数为复合性杂合子突变。突变位点分布较广,遍及整个基因的5个外显子。造成SRD5A2活性缺乏的原因:①基因点突变造成酶不能与睾酮结合。②基因缺失,接合点突变和点突变形成提前出现终止信号阻碍了正常酶分子的合成。如418delT碱基缺失,导致移码突变,提前出现终止信号,产生截短的蛋白质。③点突变影响了酶的功能(如G196S、H231R),或酶编码基因以外的突变影响了酶基因的表达。
SRD5A2缺陷症的临床表现差异很大,严重者,外生殖器为几乎正常的女性(核型46,XY),SRD5A2酶活性轻度降低者可能仅有男性外生殖器的某些缺陷或含糊,青春期时,有一定程度的雄性化,但无乳腺发育,从而促使患者由女性转向男性。
CYP19芳香化酶缺陷症
CYP19芳香化酶是催化C19类固醇激素转化为C18雌激素的最后催化酶。CYP19芳香化酶缺陷的女性有PCOS表现,男性化、女性青春期发育障碍、身材高大,血雄激素、LH和FSH升高;男性患者则表现为类阉割体型,骨骺融合延迟,高瘦身材,骨质疏松,生精功能障碍和不育学睾酮正常或升高,LH和FSH升高,而雌二醇降低。
雌激素受体缺陷症
因α-雌激素受体基因突变,患者出现雌激素抵抗综合征,男性患者表现为高身材,骨骺融合延迟,高瘦身材,骨质疏松,但外生殖器正常,血睾酮、LH、FSH正常,应用特大剂量的雌二醇亦不能促进骨骺融合延迟和骨成熟。
以上3种雄激素作用障碍与雄激素受体基因突变的临床表现有许多相似之处,其鉴别诊断见表3-16-19。
无睾症
个别男性由于遗传性或获得性因素引起无睾症(anorchia),因雌激素完全缺乏,患者没有第二性征发育,故其临床表现类似于完全型46,XY-DSD。
根据年龄/病情/意愿/疗效反应确立治疗方案
治疗方案的选择取决于诊断时患者的年龄、外生殖器畸形的严重程度、外阴对雄激素刺激的反应以及患者和家属的意愿。本病的治疗取决于患者的年龄、外生殖器畸形的严重程度以及手术纠正的难易程度和成功率。原则上应在青春发育期前作社会性别确定和外生殖器畸形矫正手术。
社会性别男性者
按男性抚养并做外生殖器整形。如果患者在儿童期确定诊断,一般应按男性性别抚养,同时应作外生殖器整形,包括将未下降的睾丸移入阴囊内、修补尿道下裂以重建阴茎尿道和关闭阴道开口。到12~13岁青春发育开始时,先给小剂量甲睾酮(methyltestosterone)含服,每天5mg,随着年龄的增大,甲睾酮剂量也逐渐增大到每天25mg。由于甲睾酮可引起胆汁潴留和黄疸,故有人认为肌注环戊丙酸睾酮(testosterone cyclopentyl propionate)或庚酸睾酮(testosterone heptanoate)为好,也应从小剂量开始,然后逐渐增加剂量。也可应用超生理剂量的睾酮,丙酸睾酮(testosterone propionate)5mg/kg,每天肌注1次;庚酸睾酮每周肌注100mg或十一酸睾酮(testosterone undecanoate)80mg/d,分4次口服。长期治疗可使血浆睾酮升高并超过成年男子正常值上限。二氢睾酮达到正常范畴,促进了患者的男性化,阴茎增大,阴毛、腋毛和胡须生长,精液量增加,性功能显著改善。大剂量睾酮治疗有一定疗效,说明2型5α-还原酶仍有少量残存的活性。超生理剂量的睾酮长期应用的有效性和安全性尚需进一步观察。
社会性别女性者
于青春期前切除睾丸并做阴道成形术。近年来,应用腔镜手术进行睾丸活检和睾丸摘除的效果很好。待到12~13岁,则开始给予雌激素替代治疗,以促进患者乳腺及其他女性特征。不管是作女性或男性抚养,治疗都应维持终生,而且患者无生育能力。
