
46,XY-DSD包括睾丸发育障碍、雄激素合成与作用障碍和一般性疾病3类。其中,睾丸发育障碍主要包括完全型性腺发育不良(complete gonadal dysplasia)、部分型性腺发育不良(partial gonadal dysplasia)、性腺退变综合征(gonadal regression syndrome)和卵-睾DSD(ovotesticular DSD)4种。
完全型性腺发育不良
以前称为完全型睾丸女性化,病因为雄激素受体(androgen receptor,AR)突变,分为完全型和不完全型两种。雄激素受体基因突变使雄激素不能与靶器官的AR结合,或虽然能结合,但雄激素-受体复合物不稳定,迅速离解而不能产生生理效应,造成雄激素不敏感,故又称雄激素不敏感综合征。AR基因定位于Xq11-12,本病属于X-连锁隐性遗传,具有家族遗传特点。
临床特点
患者的青春期出现女性第二性征发育,乳腺发育如正常女性,女性体态,但无月经来潮,阴毛和腋毛稀少或完全缺如。阴道为一盲袋,无子宫和输卵管,附睾和输精管缺如或发育不全。睾丸组织学在青春期前正常,青春期后曲细精管萎缩变小,精原细胞稀少,无精子发生,Leydig细胞结节样增生。血清LH和睾酮水平增高,FSH正常或轻度增高,E2高于正常男性,但低于正常成年女性。约10%的患者在青春期有轻度男性化改变,如阴蒂增大,阴毛和腋毛增多等。
诊断依据
染色体核型46,XY者伴女性表型和睾丸组织是性腺发育不良的基本诊断依据。青春期前完全型性腺发育不良的诊断条件是:①染色体核型46,XY,H-Y抗原阳性;②正常女性表型;③存在腹股沟疝或在大阴唇内可触及睾丸样结节,经活检证明为睾丸;④HCG兴奋试验提示存在有功能的睾丸组织;⑤阳性家族史。
青春期后完全型性腺发育不良的诊断条件是:①染色体核型46,XY,H-Y抗原阳性,AR基因突变;②女性体态,女性型乳腺发育,女性型外生殖器,盲袋阴道;③原发性闭经;④无子宫和输卵管;⑤睾丸位于大阴唇或腹股沟管内,如位于腹腔内须用B超或手术探查确证;⑥血清LH和睾酮显著增高。
治疗
性别取向取决于诊断年龄及外生殖器男性化程度。外生殖器完全女性型的患者通常作为女性抚养,青春期切除睾丸,后给予雌激素替代治疗。作为男性抚养者可行外生殖器整形(如修补尿道下裂),睾丸固定于阴囊内。幼儿期即给予小剂量睾酮(如庚酸睾酮25~50mg,每月肌注1次,3个月为1疗程)治疗,一般通过2~3个疗程可使阴茎生长达到正常同龄儿童的长度,青春期后再给予全量雄激素替代治疗。苗勒管永久综合征患者应切除苗勒管衍化器官和腹腔内睾丸,保留附睾和输精管。如果患者同时存在其他的临床异常(如高血压和低血钾等)应予以纠正。但是,给予性发育不全者性激素替代治疗时,必须观察性发育进程,并需严密追踪不良反应,因为在给予交叉性性激素(反性别性激素,cross-sex hormones)治疗后,性激素相关性肿瘤(hormonerelated tumors)的发生率增加。
注:WT1:Wilm肿瘤抑制因子-1;WAGR:Wilm肿瘤-虹膜缺失-生殖器异常- 智力低下综合征(Wilms turner, aniridia, genital anormalies and mental retardation);SF1:类固醇生成因子-1(steroidogenic factor-1);CYP17:17α-羟化酶/17,20裂链酶;3β-HSD11:3β-羟类固醇脱氢酶/△5-异构酶;17β-HSD3:17β-羟类固醇脱氢酶(氧化还原酶);DHT:二氢睾酮;AMH:抗苗勒管激素;SHBG:性激素结合球蛋白;DHEA:去氢异雄酮
部分型性腺发育不良
以前称为不完全型睾丸女性化或Reifenstein综合征。最多见的临床表现是轻度尿道下裂,尿道开口于阴茎根部,小阴茎,常伴有隐睾,无子宫和输卵管,附睾和输精管发育不良。青春期出现阴毛、腋毛生长和男子乳腺发育。睾丸小,青春前睾丸的组织学改变不明显,但青春期后曲细精管萎缩,基底膜增厚,部分透明变性,只有精原细胞,偶见初级精母细胞,无精子细胞和精子。Leydig细胞基本正常。少见的情况是患者有重度尿道下裂、盲袋阴道或只有小阴茎和分叶阴囊。激素谱和完全型性腺发育不良相似,血清LH和睾酮增高,FSH正常或轻度增高,E2轻度增高。
部分型性腺发育不良的诊断应符合以下几点:①染色体核型46,XY,H-Y抗原阳性,AR基因突变;②不同程度的尿道下裂;③男性乳腺发育;④睾丸小,阴毛和腋毛正常或接近正常;⑤血清LH和睾酮显著增高。因生殖导管和(或)外生殖器男性化不全,故需与多种疾病鉴别,如睾酮合成缺陷症、先天性肾上腺皮质增生症(类脂性肾上腺增生症3β-羟类固醇脱氢酶缺乏症、17-羟类固醇脱氢酶缺陷症)、5-α还原酶缺陷症、雄激素不敏感综合征、苗勒管抑制因子缺乏症、睾丸间质细胞发育不全、睾丸退化综合征等。
家族性46,XY型睾丸发育障碍
与Y染色体短臂性决定区或SRY基因缺失有关,又称为Swyer综合征,是X-连锁隐性或限于男性的常染色体显性遗传性疾病。Y染色体短臂缺失,SRY基因突变,常染色体基因突变或X染色体剂量敏感性反转(dosage sensitive sex-reversal,DSS)位点重复都可以引起单纯性XY型性腺发育不全。
病因和分型:
染色体核型46,XY,分为完全型和不完全型两种。Y染色体DNA杂交结果显示本病的遗传特性具有不均一性,约10%是由于Y染色体短臂性决定区缺失所致,这些患者一般有Turner综合征的躯体畸形表现。部分病例存在SRY基因部分缺失,导致转录合成的蛋白质功能丧失。此外,一些患者的分子遗传学异常是X染色体短臂包括锌指基因(ZFX)在内片段的重复所致。
临床表现:
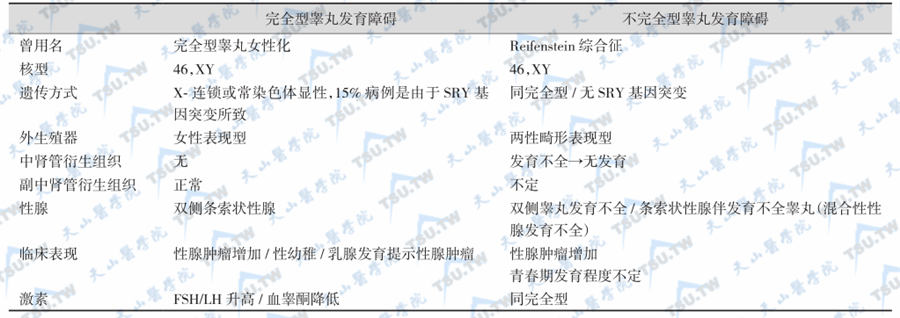
家族中受累成员的临床表现可以为完全型或不完全型(下表)两种,其差别在于性腺可以是双侧发育不全的睾丸或条索状性腺加发育不全的睾丸。生殖导管的发育依性腺而定,外生殖器为两性畸形,青春期后可有不同程度的男性化表现。完全型为女性表型,染色体核型46,XY,双侧性腺为纤维索,有子宫、输卵管和阴道,无附睾和输精管,原发性闭经。身高正常或偏高,无其他身体畸形。青春期无性发育,乳腺发育是性腺发生胚细胞瘤分泌雌激素所致。少数患者的一侧性腺为发育不全的睾丸,有部分雄激素分泌,引起阴蒂肥大,大阴唇部分融合,并有发育不全的附睾和输精管。血清睾酮水平一般高于正常成年女性。身材正常或高身材,类无睾体型,无45,XO型性腺发育不全的各种发育畸形(如性幼稚、原发性闭经、条索状性腺,有输卵管、子宫和阴道,无附睾和输精管)。血清LH和FSH增高,睾酮降低。75%患者的H-Y抗原阳性,25%为阴性。
两型家族性46,XY型睾丸发育障碍的比较
诊断:
本征的诊断要点是:①染色体核型46,XY;②性腺为纤维组织;③生殖导管衍化器官女性型或男、女两型并存;④外生殖器女性型或两性畸形;⑤类无睾丸型(eunuchoid),无其他身体畸形;⑥青春期无性发育,原发性闭经;⑦血清促性腺激素水平升高,性激素水平较男性降低;⑧SRY基因突变。
治疗:
完全型应切除双侧性腺,不完全型于矫形后行性激素替代治疗。一旦确定诊断即行预防性双侧性腺切除术。15岁左右开始给予睾酮替代治疗。诊断前已发生性腺瘤者预后不良。
