
X-性连锁先天性肾上腺发育不良症(adrenal hypoplasia congenita,AHC)主要表现为进行性精神萎靡及皮肤色素沉着,临床表现为肾上腺皮质功能减退症(adrenocortical insufficiency,ACI),患者在补充肾上腺皮质激素后可存活至成年。但进入青春期后仍无性腺发育,表现为低促性腺激素性性腺发育不良症(hypogonadotropic hypogonadism,HH)。
DAX-1为一种X-性连锁-剂量敏感性性反转-先天性肾上腺发育不良(dosage-sensitive sex-reversal-adrenal hypoplasia congenita,X-chromosome)相关性蛋白质,为核受体超家族成员之一,另一种相类似的蛋白质为类固醇生成因子-1(steroidogenic factor-1,SF-1)。这两种与AHC发病相关的调节因子主要在肾上腺皮质、下丘脑(腹中部)和垂体的LH/FSH细胞表达。DAX-1是核受体转录因子家族中的成员,一般在下丘脑-垂体-肾上腺/性腺组织中表达,主要调节肾上腺和性腺的发育、分化和激素合成与分泌功能。DAX-1基因编码一种含有470个氨基酸残基的蛋白质,突变后导致肾上腺发育不良和低促性腺激素性性腺功能减退症。DAX-1(NR0B1)和SF-1(NR5A1)是调节肾上腺和性腺发育于功能的重要因子,DAX-1突变除引起AHC外,还可能导致单一性盐皮质激素缺乏(isolated mineralocorticoid insufficiency)、性早熟(premature sexual development)和原发性肾上腺功能不全(primary adrenal insufficiency)。
DAX-1突变导致单一性原发性皮质醇缺乏症
DAX-1基因
DAX-1基因全长5kb,包含2个外显子及1个内含子。它编码含470个氨基酸残基的蛋白质,即DAX-1(下图)。DAX-1的C末端与其他核受体有同源性(可能在转录调控区)。N末端包含66~67个氨基酸重复序列,此部分与已知的DNA结合域不同,它的功能可能是与启动子的发夹样结构结合而直接调节下游基因的转录。DAX-1在肾上腺及下丘脑-垂体-性腺轴均有表达,对其发育及功能起重要作用。在小鼠,DAX-1最先出现于泌尿生殖E10.5区,这一区域在发育过程中生成肾上腺和性腺。在肾上腺,DAX-1在原囊期及最后期发育为肾上腺皮质组织;在未分化的性腺,DAX-1表达直到E12。此后,随着性腺分化的出现,DAX-1在睾丸的表达很快下降。说明DAX-1在性腺分化方面起了重要调节作用,可能是睾丸发育的一种抑制因子。雌鼠敲除Ahch基因(DAX-1)后或雌性纯合子DAX-1突变,卵巢仍能发育。尽管早期DAX-1在睾丸的表达下降,但在成熟型Sertoli细胞又再度呈高表达,提示可能在精子生成方面起关键作用。在生长发育中的前脑(E11.5)、下丘脑腹侧和腺垂体促性腺激素细胞内均鉴定出DAX-1转录子。人类DAX-1表达形式与小鼠相似,在发育中的性腺嵴、肾上腺、下丘脑、垂体、睾丸、卵巢都能检测到DAX-1的转录子。
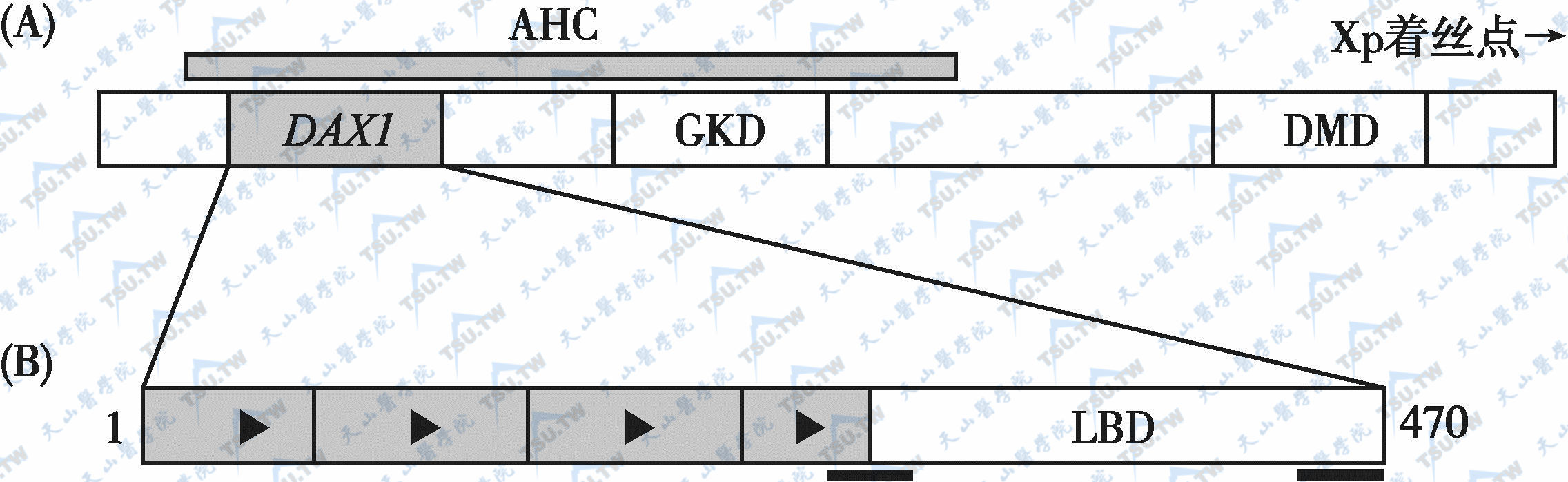
DAX1基因的结构
注:(A):DAX1的基因定位:X染色体的这一区域(Xp21)还包括编码DMD、GKD的基因。(B):DAX1蛋白:属于孤核受体超家族,C末端与其他一些核受体的配基结合域(LBD)有同源结构,而N末端则含有独特的重复序列成员。黑线条所示为假定的转录沉默区域
DAX-1突变
DAX-1基因的表达产物NROB1蛋白与下丘脑-垂体-性腺/肾上腺轴的发育有密切关系,突变型蛋白作为类固醇激素合成的负性调节因子而导致先天性肾上腺发育不良和低促性腺激素性性腺功能减退。AHC的本质为原发性肾上腺皮质功能减退,但许多患者的氢化可的松替代治疗用量较一般Addison病为高,Yeste等报道的最高用量为每日18mg/m2。AHC患者发生假性性早熟的原因与继发性ACTH过度刺激Leydig细胞ACTH受体和睾酮雄激素生成过多有关。主要为框架移位和无义突变,导致NROB1蛋白被截短。少数为错义突变、单碱基缺失或碱基插入(1个或多个)。几乎全部位于蛋白C端的保守序列中。从AHC患者中鉴定出的DAX-1基因突变类型主要有:W39X、48~49stop、Y81X、T114C、p.Q76X(c.C226T)、G498A、197stop、184stop、W236X、R267P、269delV、L278P、W291C、L295P、A300V 343delG、L381H、Y399X、405delT、E428X、I439S、457delT、L466R、I493S、501delA、R425T、629delG、656delC、702delC、728insCA、c1382-1383A ins、926-927delTG、1130delA、1141-1155del15和1464-1467del ACTC(416stop)(后两者可能为多态性,不致病)等。其中501delA既可来源于母传的线粒体DNA,又可来源于核染色体DNA。
目前已发现60余个家系的50余种DAX-1基因突变;多数是由于移码突变或无义突变导致NROB1蛋白短截。实验证明,DAX-1的C-末端缺失11个氨基酸残基就可导致肾上腺皮质功能不全和HHG。据推测,错义突变均位于配基结合区域,但I439S可能例外。
孤核受体类固醇生成因子-1异常
类固醇生成因子-1(steroidogenic factor 1,SF-1)是与DAX-1相互作用的一种因子。编码SF-1的基因(FTZF1)与DAX-1有相同的表达方式,能调节多种激素与合酶基因转录。SF-1(fushi tarazu factor-1,FTZF-1)雄性小鼠可发生肾上腺与性腺发育不全、苗勒管(Müllerian管)永存及性反转,且伴GnRH缺乏。DAX-1启动子包含SF-1结合部位,DAX-1影响SF-1介导的反式激活作用,因而DAX-1作为SF-1的抑制因子而并非激动因子而起作用,而当X连锁AHC(702delC)的DAX-1基因发生突变时,则此抑制作用消失。此外,突变型DAX-1蛋白还引起轻度肥胖和胰岛素抵抗。
野生型与突变型SF-1相互作用引起睾丸发育缺陷
DAX-1基因突变导致DAX-1功能异常,使其正常的转录抑制子SF-1的作用障碍,但患者的表型多种多样,发病年龄可早可晚,或轻或重,性腺功能障碍的程度也极不一致。其原因不明,可能与SF-1和DAX-1的修饰基因(modifier gene)的作用变异有关。SF-1是多种类固醇合酶和性发育相关基因的调节物。也是DAX-1基因转录的调节因子。DAX-1缺陷的雄性小鼠同时存在原发性睾丸发育缺陷,表现为Leydig细胞增生及生精上皮细胞的进行性退变,故DAX-1基因亦称为睾丸抑制基因。Leydig细胞中的芳香化酶(CYP19)可将睾酮转化为雌二醇,AHC患者的Leydig细胞增生,芳香化酶活性增强,由睾酮转化生成的雌二醇增多,后者再导致Leydig细胞增生和生精功能障碍。但他莫昔芬可抑制Leydig细胞增生和芳香化酶活性。Sertoli细胞表达DAX-1,对精子的发育和成熟也有调节作用。
DAX-1还有抑制其他几种靶基因(如StAR、P450scc、3β-HSD和LHβ等)活性的作用,因为DAX-1在性腺的发育过程中有拮抗SRY作用。DAX-1对肾上腺和性腺抑制的最终效果可能还受许多其他因素的影响。在青春期发育延迟的女性患者中,常可检出DAX-1基因的某些变异(如T114C、G498A多态性),这是否为特发性低促性腺激素性性腺功能减退或体质性青春期发育延迟的病因,尚需进一步研究。
突变蛋白干扰LH和FSH合成并导致下丘脑-垂体功能紊乱
近年的一些研究发现,部分AHC患者在发生AHC前即存在下丘脑GnRH和垂体LH/FSH的调节紊乱。突变型DAX-1蛋白作用于下丘脑和垂体,干扰LH和FSH的合成。Takahashi等对1例AHC男孩从出生后追踪3年,定期测定血清睾酮、LH、FSH和GnRH兴奋试验。惊奇地发现患儿3年中,下丘脑-垂体轴GnRH和促性腺激素的分泌功能是活跃的,但患者的青春前期的GnRH分泌抑制与DAX-1基因的调节有关,DAX-1基因突变则使青春期前被抑制的下丘脑-垂体-性腺轴功能不能活化,导致青春期性腺发育延迟或无发育。
其他基因变异引起AHC
有些AHC患者的DAX-1基因并无突变,其病因未明。AHC的病因可能与“基因转换”有关,即在有丝分裂期,DNA从一条等位基因转移至另一条。患者伴低促性腺激素性性腺发育不全(HHG),但卵巢的发育和肾上腺皮质功能正常。
甘油激酶缺陷症(glycerol kinase deficiency,GKD)可能是AHC的一种变异型,为一种X-性连锁隐性遗传性疾病,分为单纯型和混合型两种。单纯型的特点是致命性代谢紊乱,轻者亦可表现为“假高三酰甘油血症(pseudohypertriglyceridemia)”,患者易发生低血糖症和高酮血症,现已有数十例病例报道。混合型为Xp21邻近基因综合征(Xp21 contiguous gene syndrome)的一种表现型类型,患者还伴有AHC或Duchenne肌营养不良症(Duchenne muscular dystrophy,DMD),目前已有100多例病例报道。GKD是甘油激酶(GK)基因突变(错义突变、剪接突变或无义突变)所致。此型患者可有AHC的全部或部分临床表现。
AHC-宫内生长迟滞-骨干骺发育不良-外生殖器畸形综合征(IMAGe)可能是AHC的另一种变异型。Lienhardt 等报道7例AHC患者伴有IMAGe综合征,即表现为宫内生长迟滞(intrauterine growth retardation)、骨干骺发育不良症(metaphyseal dysplasia)和外生殖器畸形(genital anomalies),患者的DAX-1、Xp21基因串或SF-1基因均正常,这可能是另一种AHC的变异型或新的疾病综合征。
