-
为什么缺乏血管性血友病因子会有出血症状?
答:血管性血友病因子(VWF)主要由内皮细胞和巨核细胞合成,它的生成首先是合成VWF单体,然后在高尔基体中经过加工、修饰而最终形成成熟的VWF多聚体,在血浆中与FⅧ形成复合物并对FⅧ
1 -
为什么女性也会发生血友病?
答:血友病是由于F8/F9基因突变所引起的伴性隐性遗传性出血性疾病,是临床最常见的遗传性出血性疾病之一。血友病A/B(HA/HB)的遗传基因均位于X染色体上。男性患者具有一条含突变基
2 -
为什么血友病家系中的女性成员需要进行携带者基因诊断?
答:血友病A/B(HA/HB)是一种性联隐性遗传性疾病,其遗传基因位于X染色体上。血友病家系中的女性成员可能携带含有致病基因突变的X染色体但本身并无临床出血表现。但是,女性血友病携
3 -
为什么血友病需要进行基因诊断?
答:血友病A/B(HA/HB)是一种性联隐性遗传性疾病,其遗传基因位于X染色体上。男性患者具有一条含突变F8/F9基因的X染色体,不能控制FⅧ/FⅨ的正常合成,所产生的FⅧ/FⅨ分子结构缺陷或
4 -
为什么诊断血友病A要排除血友病B和血管性血友病?
答:血友病A和血友病B分别是由FⅧ及FⅨ的质或量的缺陷所导致,均属于伴性隐性遗传性出血性疾病。临床上主要表现为皮肤黏膜出血、关节出血、肌肉出血和血肿、血尿,以及出血引致的
5 -
为什么血友病表现为APTT延长而PT正常?
答:血友病分为A、B两型,分别是由FⅧ及FⅨ的质或量的缺陷所导致,均属于伴性隐性遗传性出血性疾病,也是临床最常见的遗传性出血性疾病之一,其中血友病A更常见。FⅧ的功能是作为FⅨ
6 -
为什么说冷沉淀物不是血友病A患者治疗的首选制品?
答:血友病A(hemophilia A)是凝血因子Ⅷ缺乏所导致的出血性疾病,约占先天性出血性疾病的85%,中国血友病A发病率为3~4/10万人口。由于冷沉淀在制备过程中缺乏病毒灭活,导致输注后感染
7 -
获得性血管性血友病
获得性血管性血友病(acquired von Willebrand syndrome)是一种较少见的获得性凝血系统疾病,主要继发于淋巴增殖性疾病与骨髓增殖性疾病,占48%~63%;多发性骨髓瘤与巨球蛋白血症等疾
10 -
女性血管性血友病(vWD)患者的特殊情况及处理
vWD为常染色体隐性(2型与3型)或显性(1型)遗传,男女患病几率一样。但女性因月经、妊娠与分娩等特殊情况,发生出血的机会较多,因此,在临床统计资料上女性患者占60%以上。大约65% 的vWD
11 -
血管性血友病(vWD)的治疗
血管性血友病(vWD)患者应尽量避免创伤与手术,避免运用影响血小板功能的药物如阿司匹林、吲哚美辛与低分子右旋糖酐。对有局部轻微创伤、鼻出血与牙龈出血者可用明胶海绵填压。
12 -
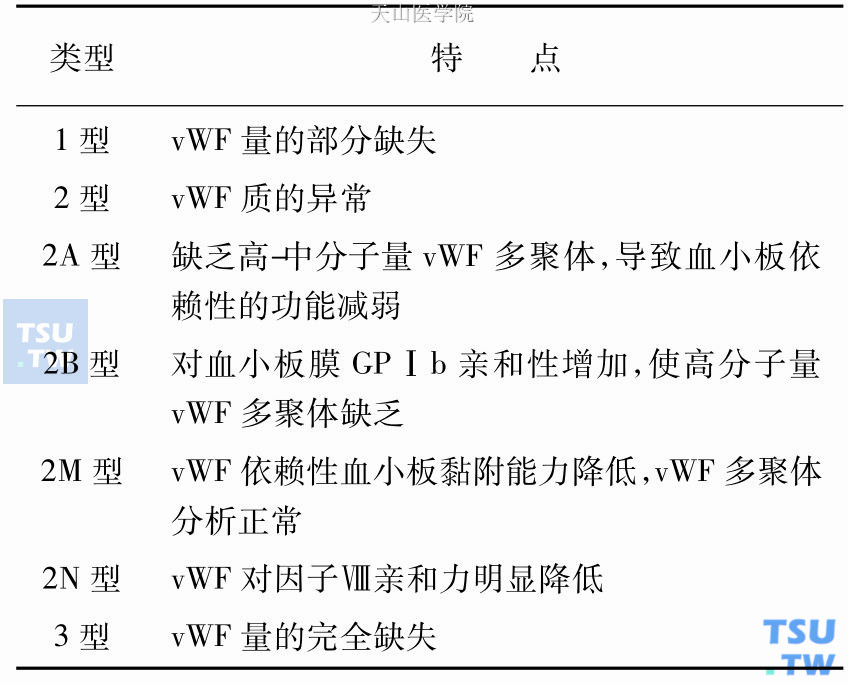
血管性血友病(vWD)的分型
vWD可以是vWF量的减少,也可以是质的异常,其临床表现及实验室检查结果很不相同。国际血栓与止血学会vWF委员会根据vWD的发病机制与表型提出的分型方法并被我国第六次血栓与止血
13 -
血管性血友病(vWD)临床表现与实验室检查
vWD多为常染色体显性遗传,少数为常染色体隐性遗传,男女均可患病。患者有皮肤黏膜出血的倾向,以鼻出血与牙龈出血最常见,这与血友病以关节及软组织出血为主的临床表现有很大不同
14 -
血管性血友病因子(vWF)分子结构及病理生理意义
vWF是一种重要的血浆成分,其分子结构已经阐明。cDNA测序证实vWF的mRNA长度为8. 7kb,其开放阅读编码含2813个氨基酸的蛋白质,包括22个氨基酸的信号肽,741个氨基酸的前导肽与2050
15 -
关于血管性血友病(vWD)
血管性血友病亦称为von Willebrand病(von Willebrand disease,vWD),于1926年由芬兰医生Eric von Willebrand首先报道。其发现一个家系男女多人有明显的皮肤黏膜出血,但深部组织出
16 -
遗传性因子Ⅺ缺乏症(血友病C)
遗传性因子Ⅺ缺乏症由Rosenthal等首先报道于1953年,当时命名为血友病C,但现在一般称为因子Ⅺ缺乏症。本病多见于犹太人后裔,其杂合子发生率可达2%~13%,纯合子发生率约0. 1%。但在
17 -
遗传性因子Ⅴ缺乏症(副血友病)
Owern于1947年首先报道此病。此病有时被称为副血友病。在凝血因子Ⅴ(FⅤ)发现前,曾认为有一个加速因子(又称易变因子,曾被命名为凝血因子Ⅵ),以后的研究证明前加速因子和因子Ⅴ为同
18 -
获得性因子Ⅸ抑制物
血友病B(hemophilia B)患者接受因子Ⅸ浓缩物治疗后较少出现因子Ⅸ抑制物,抑制物的发生率为3%~5%。与血友病A类似,严重因子Ⅸ缺乏的血友病B患者产生因子Ⅸ抑制物的可能性增加,发生
19 -
获得性vWF抑制物
与血友病A相似,遗传性vWF缺乏所致血管性血友病(hereditary von Willebrand disease,vWD)患者,在接受替代性输注治疗后可以产生vWF抑制物(抗vWF抗体)。在非遗传性血管性血友病患者中
20