
调控输精管道的基因
有关输精管道调控基因,目前许多问题尚不明确。相关研究多为附睾基因。
附睾特异基因的研究也是近10几年才开展的工作,目前已被克隆出来在附睾中表达的附睾特异基因不超过15种,而人类附睾特异基因目前只有5种。目前对于附睾特异基因的研究主要在美国和德国的实验室开展。近年来,中国科学院上海生物化学研究所张永莲和刘强等在猴身上获得了11个未知附睾基因并进行了克隆,还对附睾区域分布和雄激素调控进行了研究,最终发现SC-342、SC-177、SC-112、SC-165这4个基因和人类同源,并且SC-342是附睾高度特异的,SC-177在人附睾中含量丰富。目前已了解的附睾特异基因主要见下表。
附睾特异基因相关情况汇总表
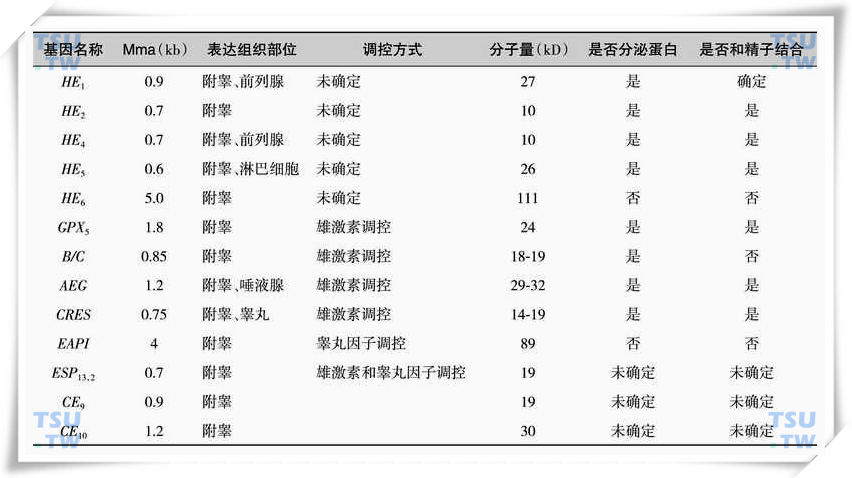
附睾基因表达有很多特点,主要表现为:①高度的组织特异性:这些基因主要在附睾表达,在其他组织中鲜有表达或表达很低。②高度的区域性:不少基因,如B/C、HE2、EAPI、GPX5、CRES、SC-384、SC-531等,仅在附睾头部表达,尽管精子成熟(运动能力和受精能力的发育)主要在附睾体部,但是附睾头部作为蛋白合成和分泌非常活跃的区域,在调节角度发挥的作用更大。当然HE1、HE4、 HE5、SC-177、SC-461等主要在附睾体部和尾部表达,也有少部分基因在附睾头-体-尾全长表达。③高度的细胞特异性:许多蛋白及mRNA在附睾同一区域表现出交叉排列的表达特征,在一些主细胞表达强烈,而其相邻的主细胞则表达量很低,这就反映了这些上皮细胞尽管形态相似,但是却可能处于不同的功能状态或具有不同的功能。
附睾基因表达受到雄激素、附睾因子和睾丸因子的调控。这些调控机制的变化都会对与之相关的基因产生影响。
输精管道先天性发育异常及其相关基因
二、MRKH综合征(Mayer-Rokitansky-Küster-Hauser syndrome,MRKH syndrome):
先天性无阴道、无子宫或始基子宫,输卵管、卵巢及外生殖器发育正常,称之为Mayer-Rokitansky-Küster-Hauser syndrome(MRKH)综合征。正常情况下女性胚胎发育过程中副中肾管头端发育成输卵管,中、尾端发育成子宫及阴道上段。双侧副中肾管中、尾段发育时相互融合,纵隔消失,形成单一的子宫阴道腔。MRKH综合征患者双侧副中肾管中、尾段早期发育时未能融合即停止发育,形成双始基子宫,阴道上、中部闭锁。近年研究认为发生MRKH综合征系基因缺陷所致,相关基因包括WNT4、HOXA7-13、PBX1、WT1、PAX2等。
WNT4在肾发育中起重要作用。Biason-Lauber等人对一位MRKH综合征患者研究发现,该患者存在WNT4基因突变。在生殖道的发育中,HOX基因家族成员HOXA7-13的表达缺陷可能导致MRKH综合征。此外,在早期发育中发挥作用的基因如WT1,PAX2等,也可能是MRKH综合征的候选基因,尽管其具体作用尚不明确。
一、先天性输精管缺如(congenital absence of the vas deferens,CAVD):
多为双侧或单侧完全缺如,包括阴囊段和腹股沟段缺如(外缺如)与盆腔段缺如(内缺如)。囊性纤维化病(cystic fibrosis,CF)是外分泌腺的分泌功能紊乱,可有呼吸道和消化道的异常,在男性生殖道方面常存在附睾的分泌障碍,造成渐进性的精道阻塞,引起输精管闭锁或缺如、附睾结构异常和精囊腺缺如。与CF相关的CFTR基因突变所致的中肾管再通障碍可能是CAVD发病的重要原因。
