
糖尿病、高脂血症和心血管系统疾病(高血压、冠心病等)的发病率逐年上升,已成为影响人们生活质量和引起死亡的重要原因。国内外许多大规模的研究,如4S、UKPDS、HAPPY及HOT等都强调长期用药的重要性和必要性。但长期使用药物不可避免地会带来一些副作用,如对血糖、血脂代谢的影响。因而在治疗一种疾病的同时,又可能成为另一种疾病发生的隐患。
目前引起人们关注的主要是抗高血压和冠心病等心血管用药对血糖、血脂代谢的影响。这类药物主要包括利尿剂、β受体阻断剂、钙拮抗剂、血管紧张素转换酶抑制剂(ACEI)、α受体阻断剂、β受体阻断剂。
利尿剂
在20世纪70年代,利尿剂曾是治疗高血压的首选药物并被广泛使用,然后才考虑诸如β受体阻断剂等抗高血压药。虽然现在治疗高血压的药物治疗方案有了很大变化,但利尿剂仍然是治疗高血压的重要药物和治疗心功能不全的常用药物。利尿剂是一类促进体内水分和电解质(Na+为主)排出的药物,通过影响肾小球的滤过率、肾小管的重吸收和分泌等功能而实现其利尿作用。
分类
常用的利尿药依据其作用部位、化学结构及作用机制分为以下四类:
- 主要作用于髓袢升支髓质部的利尿剂(袢利尿剂),如呋塞米(furosemide,速尿),依他尼酸(ethacrynic acid,利尿酸)等,属于高效利尿剂。
- 主要作用于髓袢升支皮质部的利尿剂,噻嗪类(thazide),氯噻酮(chlorothalidone)等,为中效利尿剂。
- 主要作用于远曲小管的利尿剂,螺内酯(spironolactone,为醛固酮抑制剂)、氨苯蝶啶(triamterene)等,为保钾利尿剂。
- 主要作用于近曲小管的利尿剂,如乙酰唑胺(acetazolamide)等碳酸酐酶抑制剂。
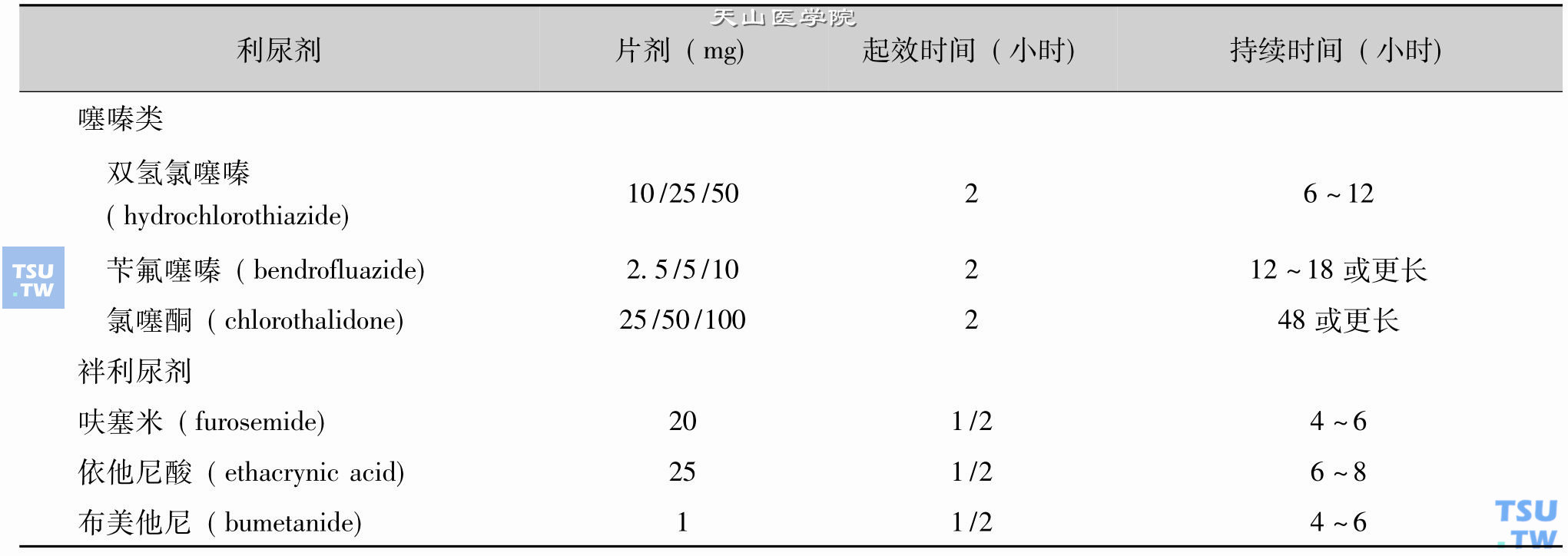
大量研究表明,对代谢影响较大的主要为前两类利尿剂。这两类利尿剂种类较多,口服后起效及作用时间有较大的差异,对代谢影响的程度与药物的剂量、作用时间和使用时间的长短有一定的关系,详见下表。
部分利尿剂的临床特点
对代谢的影响
1)血糖升高和胰岛素敏感性下降:噻嗪类利尿剂从1957年开始应用至今,在治疗高血压、心功能不全等方面的作用被普遍肯定和认同,仅在美国就有超过五百万患者在使用这类药控制血压。但长期使用引起葡萄糖耐量异常和胰岛素敏感性下降,使患糖尿病的危险性增加以及加重糖尿病糖代谢紊乱,越来越受到人们的重视。这类利尿剂使用的剂量和时间与代谢副作用的产生和损害程度密切相关。
Steven G等人在一项研究中给两组患者分别服用氢氯噻嗪12. 5mg/d和25mg/d。治疗后4周,血糖水平无明显改变,但到12周时每日服用25mg的患者血糖水平显著升高(P<0. 01),并伴有血钾下降。但两组的降压效果却近似,分别使舒张压下降44. 3%和46. 8%,无显著性差异。也有人研究发现给高血压患者服用氯噻酮50mg/d,八周后血糖和血胰岛素水平都显著上升;血糖与血钾浓度呈正相关,与血镁呈负相关;红细胞内K+及Mg2+水平与血胰岛素水平呈正相关。此外还有报道利尿剂可以影响质膜对葡萄糖的运输。在基础状态下,葡萄糖的跨膜运转主要依靠葡萄糖运载体-1(GLUT1),但在胰岛素分泌水平上升时,GLUT4起主要作用。呋塞米(furosemide)和氢氯噻嗪可使大鼠脂肪细胞对胰岛素介导的葡萄糖转运能力下降,可能就与GLUT4活性下降有关。临床研究还发现,袢利尿剂对糖代谢的影响远小于噻嗪类利尿剂,但大量及长期使用时对糖代谢的影响仍不能忽视。一些临床研究观察到每日服用一次利尿剂要比同样剂量分两次口服对代谢的影响小。有文献报道苄氟噻嗪(bendrofluazide)1. 25~2. 5mg/d能有效控制血压,而不会引起糖代谢异常。
螺内酯对糖代谢基本上没有影响。但服用一段时间后可以引起血容量下降和血钾升高,因此应引起注意。
利尿剂对糖代谢影响的可能机制有:①利尿剂引起的血清钾、镁及细胞内钾、镁代谢紊乱使外周组织细胞对胰岛素的敏感性下降,这些组织对葡萄糖的摄取能力减弱;②可能存在因低钾血症抑制胰岛β细胞分泌胰岛素的作用。
总之,利尿剂在代谢方面的副作用与药物种类、代谢时间、剂量、服用次数、使用时间有关。但可以肯定的是袢利尿剂所导致的副作用远低于噻嗪类利尿剂。这些方面在临床用药中都应注意。
2)血脂异常:噻嗪类和袢利尿剂等排钾性利尿剂都可以使血脂代谢异常。有文献报道,总胆固醇上升4%~13%、LDL胆固醇上升7%~29%、VLDL胆固醇上升7%~56%及甘油三酯上升14%~37%。但这些代谢异常在停药后均可恢复。
Middeke M等人研究发现高血压患者服用氢氯噻嗪(平均服用剂量33mg/d)和卡托普利(平均服用剂量64mg/d)12个月后,甘油三酯升高22. 5%、HDL胆固醇降低6. 1%。
吲达帕胺为甲基二氢吲哚(methylindoline)类药物,也有血管扩张的作用,不会引起血脂异常,每日服用2. 5mg安全、有效。因此,比较适合于高血压患者服用。保钾性利尿剂对血脂基本上无不良影响。
α肾上腺素能受体拮抗剂
分类
α受体可分为α1和α2两种亚型。根据这类药物对两种受体的选择性可分为:
- α1、α2受体阻断剂:酚妥拉明(phentolamine)、妥拉唑林(tolazoline)和酚苄明(phenoxybenzamine);
- α1受体阻断剂:哌唑嗪(prazosin)、特拉唑嗪(terazosin,或称高特灵)和多沙唑嗪(doxazosin);
- α2受体阻断剂:育亨宾(yohimbine)等。
α受体拮抗剂是治疗高血压的有效药物之一。但它的降压效果主要取决于对血管平滑肌上α肾上腺素能受体中α1亚型的选择性。阻断α1受体可以使动脉扩张,降低血管的张力。研究发现在血管内皮细胞上还存在肾上腺素能受体α2亚型,阻断这一受体后引起内皮细胞依赖性扩张,而选择性α1受体拮抗剂不产生这种效应。
目前临床上常使用的α受体拮抗剂有哌唑嗪(prazosin)、特拉唑嗪(terazosin,或称高特灵)和多沙唑嗪(doxazosin)。它们的作用机制和降压效果相似,但药物作用时间和使用剂量却不同。哌唑嗪应用的最早,起效快,口服后1~3小时达到峰值,半衰期为2~3小时,因而每日需服用2~3次;特拉唑嗪口服后2~3小时达峰值,半衰期为12小时,每日服用1~2次;多沙唑嗪在服药后2~6小时作用最强,半衰期可达22小时,故每日服用一次即可。
对代谢的影响
α1受体拮抗剂对高血压患者的代谢水平基本上无不良影响,1994年的一项研究发现多沙唑嗪(doxazosin)还能改善葡萄糖代谢及降低胰岛素水平。α1受体拮抗剂对糖代谢的影响可能是通过体循环动脉扩张,肌肉中血流量增加,有利于组织对葡萄糖及胰岛素的利用来实现的。α1受体拮抗剂是抗高血压药中少有的一种能改善脂代谢的药物,单独使用特拉唑嗪(terazosin,或高特灵)可降低总胆固醇(TC)5%、甘油三酯(TG)6. 1%和低密度脂蛋白-胆固醇(LDL-C)7. 6%,但高密度脂蛋白-胆固醇(HDL-C)无改变。该药分别与利尿剂、β受体拮抗剂、钙拮抗剂或ACEI合用降低TC 4. 3%~5. 4%和TG 4. 8%~6. 3%。此外,这类药物不会引起血尿酸的升高。
胰岛β细胞膜上有α肾上腺素能受体,按其生理效应、药理特性和分布部位的不同可分为α1受体和α2受体。α2受体可进一步分为α2A受体和α2B受体。α2A受体兴奋有抑制胰岛素分泌的作用。选择性的α2A受体拮抗剂咪唑克生(idazoxan)及非选择性的α2受体拮抗剂酚妥拉明、育亨宾能拮抗α2A受体受体兴奋剂引起的对胰岛素释放的抑制。
β肾上腺素能受体拮抗剂
β受体拮抗剂与利尿剂是心血管系统用药中对代谢水平,主要是血糖和血脂影响最大的两种。选择性β受体拮抗剂的影响要小于非选择性β受体拮抗剂,这些药物引起的代谢异常在停药后可以完全恢复。
分类
β受体可分为β1和β2两种亚型。因此,β受体阻断剂又可分为:①β1、β2受体阻断剂:普萘洛尔(propranolol,心得安)、噻吗洛尔(timolol)、吲哚洛尔(pindolol)、索他洛尔(sotalol)及阿普洛尔(alprenolol)等;②β1受体阻断剂:阿替洛尔(atenolol,氨酰心安)、美托洛尔(metoprolol)等。这些药物中,有些还具有较弱的激动β受体的作用,即内在拟交感活性,所以,上述两类药物可再分为无内在拟交感活性者和有内在拟交感活性者。β受体阻断剂的作用特点详见下表。
β受体阻断剂的分类及作用特点

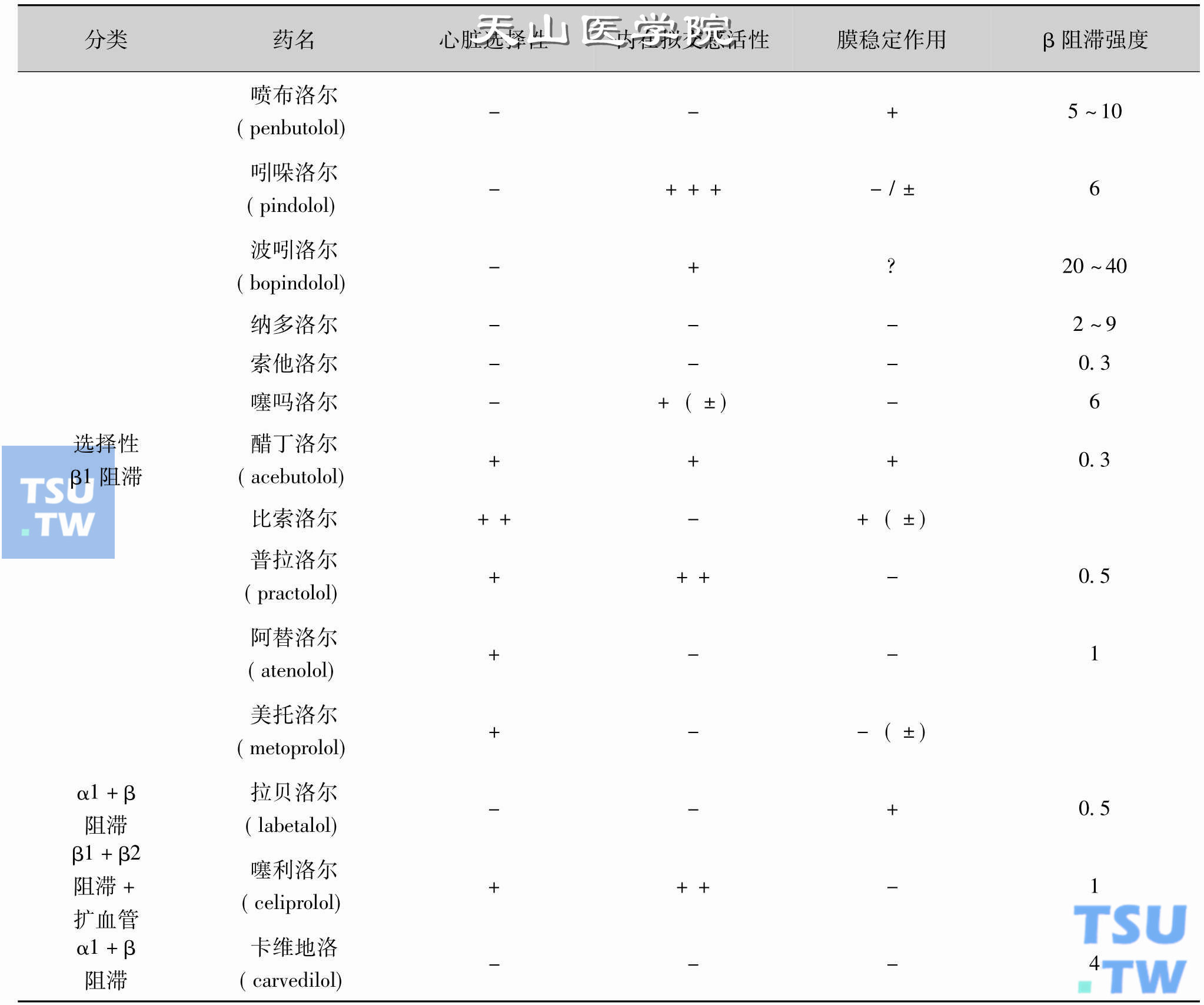
(摘自:陈修,陈维洲,曾贵云.心血管药理学.第2版. 1998:167-168)
对代谢的影响
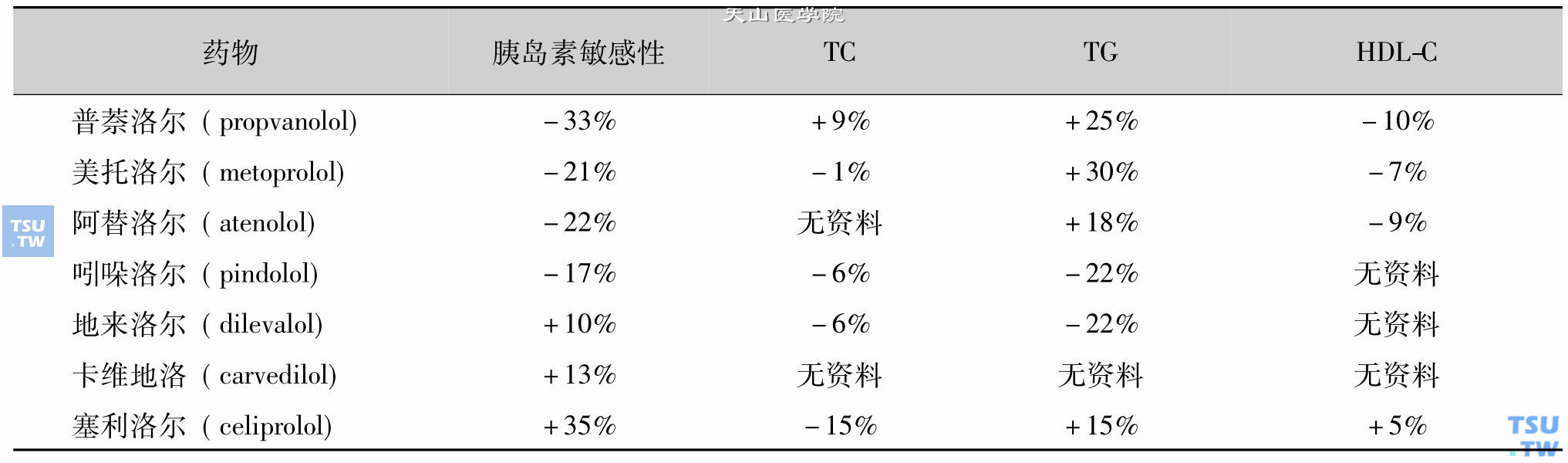
Lars Lind等人对使用β受体拮抗剂的高血压患者进行了两年的观察,发现阿替洛尔(atenolol)150mg/d、美托洛尔(metoprolol)200mg/d、吲哚洛尔(pindolol)50mg/d及普萘洛尔(propranolol)160mg/d,对血糖及血脂代谢水平均有影响。普萘洛尔及吲哚洛尔使用半年后引起空腹血糖上升及葡萄糖耐量减低,这些变化一直持续到观察结束(24个月)。此外,患者在使用普萘洛尔、阿替洛尔及美托洛尔6个月后空腹胰岛素水平上升,胰岛素分泌延迟。对血脂影响方面,选择性或非选择性β受体拮抗剂在使用4~6个月时均可以使HDL-C下降,吲哚洛尔使TC上升,普萘洛尔、阿替洛尔及美托洛尔可引起VLDL和TG水平上升。
β受体拮抗剂对代谢影响的机制:
- 抑制胰岛素第一时相的分泌:胰岛素分泌的第一时相对血糖控制非常重要。而胰岛素分泌水平的下降必然引起第二时相高分泌。因此,第一时相的胰岛素分泌受到影响往往是2型糖尿病发病的重要因素。
- 胰岛素敏感性下降:高血压患者存在不同程度的胰岛素抵抗。β受体拮抗剂可以使体内胰岛素清除能力下降,进一步加重了高胰岛素血症,最终使靶细胞上的胰岛素受体下调降低胰岛素的敏感性。如下图所示。
- 另外,β受体拮抗剂所引起的血流动力学改变可能也是一个原因。β受体拮抗剂可以使体重上升及心输出量下降,外周组织血流灌注受到影响而减少,组织细胞摄取和利用的葡萄糖量减少,使血糖上升,引起高胰岛素血症。心输出量下降还干扰了葡萄糖氧化供能机制,继发性引起生长激素水平升高,对血糖都有影响。
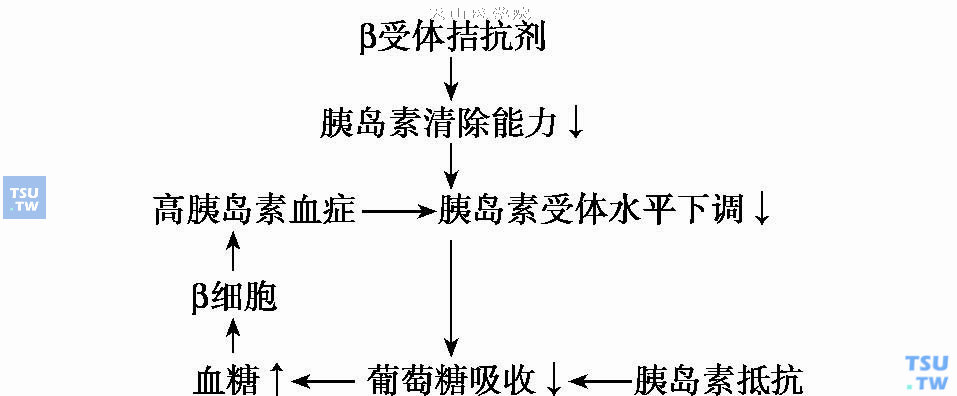
β受体拮抗剂对糖代谢的影响
有人认为β2受体激动剂可使血管扩张,而非选择性β受体拮抗剂会阻断了这一受体,对代谢的影响比选择性受体拮抗剂要大。但这一假设未得到证实。临床研究发现,同时阻断α1受体的非选择性β受体拮抗剂如地来洛尔(dilevalol),卡维地洛(carvedilol)和塞利洛尔(celiprolol)不仅未影响胰岛素敏感性,反而有轻度的改善作用。但这些现象还有待于进一步的研究来证实。地来洛尔因副作用已较少使用。部分β受体拮抗剂对胰岛素敏感性和血脂代谢的影响见下表。
β受体拮抗剂对代谢水平的影响
钙拮抗剂(CCB)
分类
细胞内钙水平的调节是通过以下途经实现:钙离子通道(受体调控的和电压依赖性通道)、钠钙交换、钙泵以及细胞内部的钙摄取与释放(线粒体和肌浆网等)。现有的CCB都是作用于电压依赖性的钙通道。目前已知的电压依赖性的钙通道有6种:L、T、N、P、Q和R型,后四种主要与神经细胞有关。L(long lasting)型通道介导长时间的钙离子转运,并且失活缓慢。目前的三类主要CCB都是抑制L型通道的。
这三类CCB为:①双氢吡啶类(DHB),以硝苯地平(nifedipine,心痛定,nifehpine,拜新同),氨氯地平(amlodipine,络活喜),拉西地平(lacidipine,乐息平),非洛地平(felodipine,波依定)为代表;②苯噻嗪类,以地尔硫䓬(合心爽)为代表;③苯烷胺类,以维拉帕米(verapamil,异博定)为代表。这几类CCB作用于L型通道的不同部位,药代动力学特点以及对不同组织的选择性也不同。
对代谢水平的影响
钙拮抗剂、利尿剂、β受体拮抗剂、ACEI以及α受体拮抗剂是治疗高血压的一线药物。许多研究表明β受体拮抗剂和噻嗪类利尿剂对血糖、血脂代谢均有影响,Framingham Cohort发现血糖从3. 9~4. 4mmol/L(70~79mg/dl)上升到5. 0~5. 5mmol/L(90~99mg/dl)时,心血管疾病的发病率男性增加11%,女性增加21%。Francesca等人的研究表明,高血压患者和糖尿病合并高血压患者分别服用氨氯地平5~10mg/d共8个月,治疗前后血糖、血脂代谢水平及胰岛素敏感性均无显著改变。此外,在对糖耐量正常和糖耐量异常的高血压患者的观察中发现,服用非洛地平5~9mg/d 7. 5个月后糖代谢指标未受影响,而糖耐量正常的患者HDL-C和ApoA-I水平显著下降,两组TC、TG、LDL及apoB水平均无显著改变。国内学者研究发现,胰岛素敏感性减低的高血压患者,氨氯地平5~10mg/d治疗4周后葡萄糖代谢率明显提高,而胰岛素敏感性正常的高血压患者葡萄糖代谢率无明显增加,说明氨氯地平可以明显改善胰岛素减低的高血压患者的胰岛素敏感性。其机制可能是氨氯地平有效地抑制心肌和血管平滑肌的钙离子跨膜内流,扩张血管平滑肌和增加血流量,改善了周围组织对葡萄糖的利用。也有人报道,2型糖尿病患者在服用硝苯地平后基础血清血糖、胰岛素、C肽和胰高糖素水平以及馒头餐负荷后各时点血糖及峰值无改变,30分钟及60分钟时的胰岛素、C肽水平亦无显著改变,但120分钟时的血清胰岛素水平明显下降,C肽的测定结果与胰岛素相符,表明硝苯地平抑制了餐后120分钟峰期时的胰岛素分泌。各时段的胰高糖素水平则无改变。硝苯地平引起糖代谢异常的最低剂量是20mg/d,最短用药期限为3~10天。但也有文献显示药物剂量即使达到60mg/d或延续3个月也未发现对血糖的影响。
曾经有报道,钙拮抗剂抑制胰岛素分泌,使糖代谢紊乱。1966年,Godsky和Bemmett报道,用不加钙的含葡萄糖液灌注离体大鼠胰腺没有产生胰岛素分泌;1973年Malaisse等观察到钙拮抗剂D600(戈洛帕米,gallopamil)有剂量相关性抑制离体胰岛细胞的Ca2+摄取和胰岛素释放。维拉帕米、地尔硫䓬、尼卡地平等钙离子拮抗剂对胰岛素的分泌也有抑制作用。钙离子在许多代谢机制中发挥重要的作用。胰岛β细胞受葡萄糖诱导的胰岛素分泌包括两个时相,第一时相分泌不依赖于细胞外钙,因而不被钙拮抗剂所抑制;第二时相分泌与钙内流增加有关。所以,钙拮抗剂可能主要影响第二时相时的钙内流抑制胰岛素的分泌。动物研究发现硝苯地平和维拉帕米可引起可逆性、剂量依赖性胰岛素分泌抑制。1990年,Westfall等用比目鱼肌研究发现,低浓度的钙激动剂(BAK8644)能提高胰岛素介导的骨骼肌葡萄糖转运,而这一作用可被硝苯地平所抑制。但动物实验的研究结果在临床研究上报道不一,基本上未得到充分的证实,这可能和实验中使用的药物剂量远远大于临床用药剂量有关。
血管紧张素转换酶抑制剂
血管紧张素转换酶抑制剂(ACEI)最初是作为降压药研制的。ACE是一种二价二肽羧基金属肽酶,存在于内皮细胞、上皮细胞、神经上皮细胞和脑中,以及血液和许多体液中。ACE分解血管紧张素Ⅰ(ATⅠ)和缓激肽的羧基末端肽,调节肾素-血管紧张素系统(RAS)和缓激肽-激肽系统。ATⅡ是一个强血管收缩剂,直接作用于血管平滑肌细胞,在外周和中枢与交感神经系统相互作用增加血管张力;也能通过醛固酮、肾血管收缩及抗利尿激素机制引起水钠潴留;在细胞水平促进细胞移行、增生和肥厚。ACE调节缓激肽扩张血管、利尿作用与ATⅡ缩血管和钠潴留作用之间的平衡。而ACEI通过降低ATⅡ的生成和缓激肽的降解来发挥治疗作用。
分类
国内外临床使用的ACEI超过10种,它们在活性基团的化学结构、效力、生物利用度、血浆半衰期、排泄途径、分布、对ACE的亲和力方面均有所差别。根据活性基因的化学结构可以将ACEI分为三大类:①含有巯基的一类;②含磷酸基团的一类;③含羧基基团的一类,详见表74-4。这类药物中福辛普利、群多普利和螺普利经肾脏和胆道双通道排出,其他ACE抑制剂均通过肾脏排出。国内外使用的ACEI结构与作用特点详见下表。
ACEI的结构与作用特点
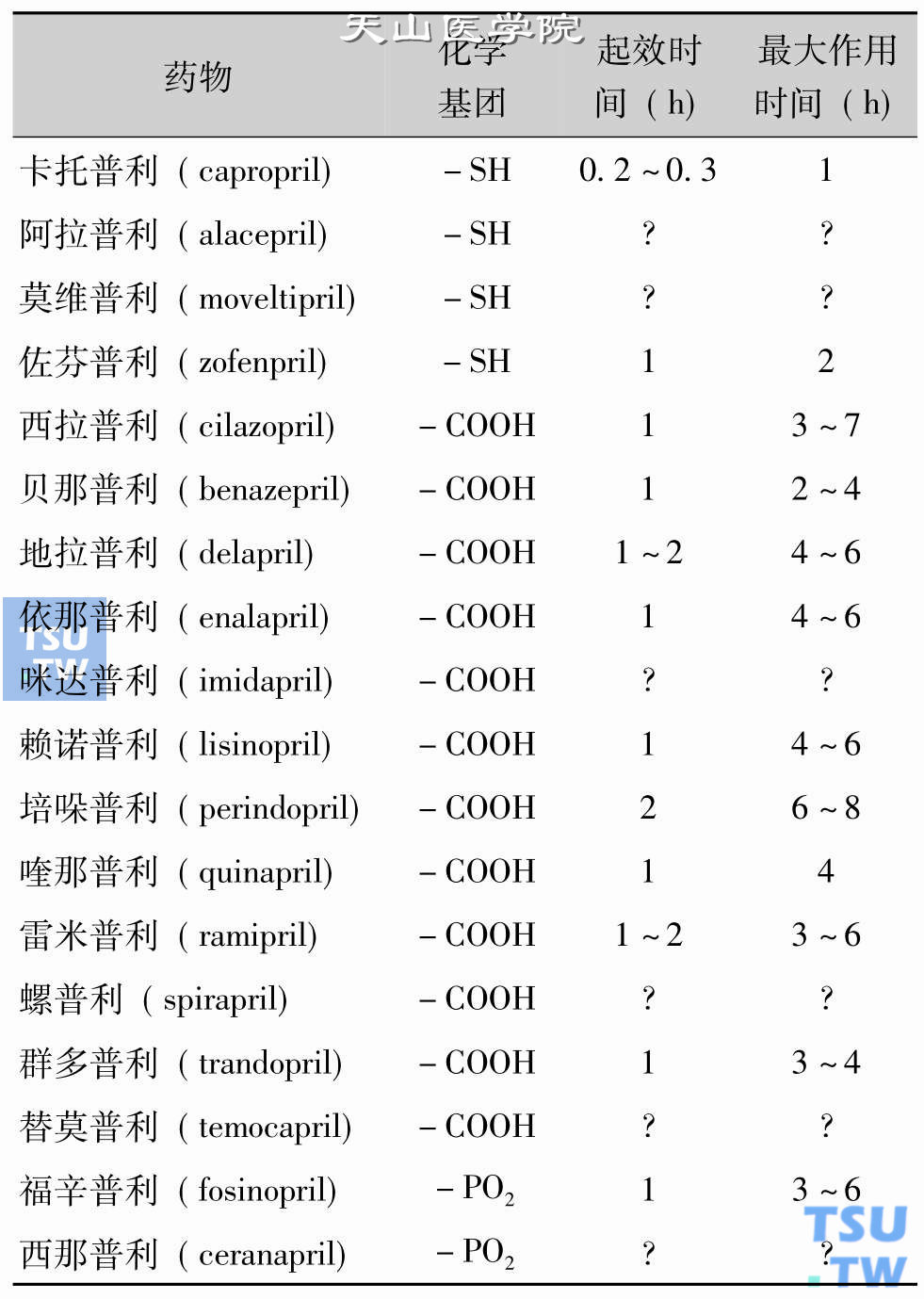
ACEI分子中的功能基团有3种:巯基、羧基及磷酸基。功能基团是与靶酶(ACE)分子中锌(Zn)结合的关键部位。近年来发现,不同功能基团的药物对组织的选择性不同。巯基类对心脏的选择性较高,羧基对肾脏的选择性较高,而磷酸基则对脑组织的选择性强。另外,含羧基的ACEI有改善胰岛素敏感性作用,其他两类作用相对较小或无作用。
对代谢的影响
ACEI对代谢的影响一直是医生关注的一个问题。许多研究表明,依那普利对糖代谢的改善作用较明显。其作用机制为:①改善骨骼肌和脂肪组织葡萄糖的摄取,增加胰岛素介导的葡萄糖的利用,提高胰岛素的敏感性;②减少肝葡萄糖生成及胃肠道对葡萄糖的吸收。
血管紧张素受体拮抗剂
ACEI的某些副作用,如咳嗽等在很大程度上影响了这一类药物的应用,因而进入20世纪90年代,血管紧张素Ⅱ(ATⅡ)受体拮抗剂在临床上逐渐应用并被认为是有前途的药物。
血管紧张素Ⅱ受体有四种亚型,即AT1、AT2、AT3和AT4。AT1和AT2主要存在于人类的大多数组织中,但作用不同。AT1受体是一种膜受体,它具有受体的所有特征,介导血管紧张素Ⅱ受体的几乎所有功能。血管紧张素Ⅱ受体拮抗剂对AT1受体有三种拮抗方式:竞争性、非竞争性和中间型(混合性)。血管紧张素Ⅱ受体拮抗剂化学分类为:
- 联苯四唑类,氯沙坦(losartan,中间型),坎地沙坦(candesartan,非竞争性),厄贝沙坦(irbesartan)及他索沙坦(tasosartan,竞争性);
- 非联苯四唑类,依普罗沙坦(eprosartan,竞争性)和替米沙坦(telmisartan,中间型);
- 非杂环类,缬沙坦(valsartan,中间型)。
有研究表明,该类药物对血糖和血脂的影响不大,但仍有血钾增高的作用,需要进一步的验证。
