
肿瘤恶液质发病机制的三个中心环节是:摄食减少(厌食)、体重丢失、肌肉减少。
厌食(摄食减少)
恶液质是一种代谢异常综合征,这种代谢当然包括能量代谢,突出地表现为能量摄入不足及能量消耗增多。图4-3-1说明摄食的生理调节系统。
厌食引起的食欲下降、进而导致的能量摄入减少毫无疑问是肿瘤恶液质患者体重丢失的重要因素。但是,厌食如何发挥作用还不清楚,是独立发挥作用还是通过炎症反应发挥作用还不知道。厌食由很多组成成分如恶心、味觉改变、吞咽困难、抑郁等组成。激进的营养支持不能逆转体重丢失说明厌食导致的营养不足不是肿瘤恶液质的始动因素,而厌食可能是恶液质的一个后果,所以,有人认为食欲下降是肿瘤因子或宿主对肿瘤免疫反应的造成的一个次级反应。细胞因子可以抑制神经肽Y(neuropeptide Y,NPY)通路或模拟瘦素对下丘脑的负反馈作用,从而导致厌食。在一组胃/食管癌(n=220)的研究中发现,83%患者有体重丢失,多因素回归分析发现饮食摄入、血浆CRP浓度及肿瘤分期是体重丢失的独立影响因素,三者的影响力为38%、34%、28%,说明恶液质患者的体重丢失不仅仅是由于能量摄入降低所致。下丘脑弓状核有两组神经元调节食欲:黑皮质素系统(melanocortin system)及NPY系统。NPY直接刺激食欲或通过释放其他食欲刺激蛋白刺激食欲,黑皮质素系统神经元释放α黑素细胞刺激激素(α-melanocytestimulating hormone,α-MSH),并通过黑皮质素3,4受体(melanocortin-3 and 4 receptors,MC3R,MC4R)导致觅食行为(food-seekingbehaviour)抑制,增加基础代谢率,瘦体组织减少。刺鼠相关蛋白(agoutirelated protein,AgRP)是NPY释放神经元释放的另外一种神经肽,它与MC4R-刺激蛋白提高食欲的作用起对抗作用。这些食欲神经元还为循环中的瘦素及IL-1β表达受体,而瘦素及IL-1β两者均下调食欲及食欲刺激蛋白脑肠肽(ghrelin)受体。
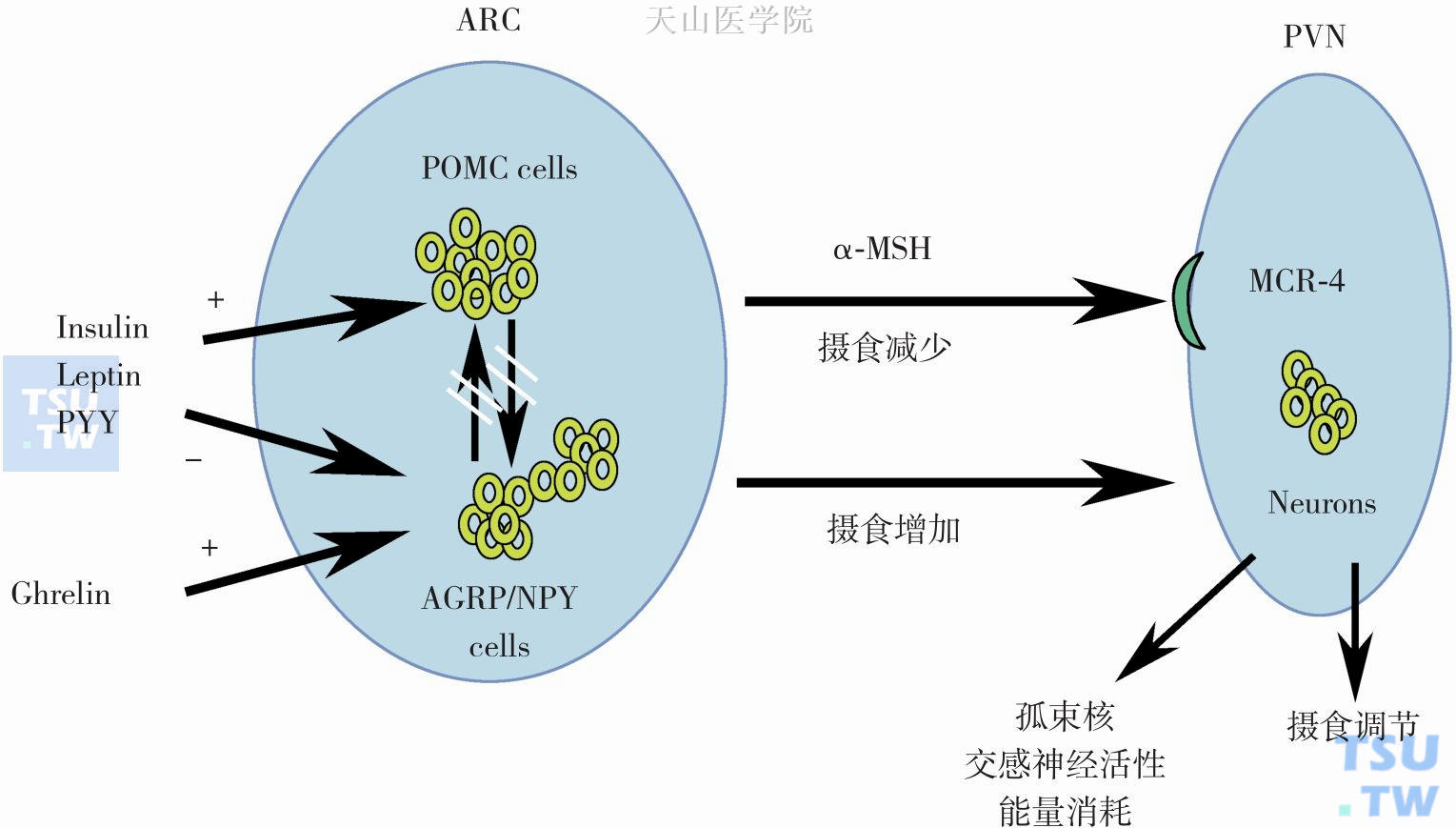
图4-3-1 摄食神经内分泌调节系统
外周的长期(胰岛素、瘦素)和短期(酪酪肽、食欲刺激素)信号调节下丘脑弓状核合成饥饿(刺鼠相关蛋白和神经肽Y)和饱足神经肽(前阿片黑素细胞皮质激素),饥饿及饱足神经肽作用于下丘脑室旁核以调节食物摄取。图中ARC:下丘脑弓状核;PVN:室旁核;NPY:神经肽Y;MCR-4:黑皮质素受体-4;AgRP:刺鼠相关蛋白;POMC:前阿片黑素细胞皮质激素;α-MSH:α-促黑激素;PYY:酪酪肽;中断的箭头:活化的POMC神经元抑制AgRP/NPY神经元,反之亦然
体重丢失
人们对不同条件下体重丢失及身体组成变化的认识也是一个变化过程,而且时至今日,认识也未完全统一。
2009年Tisdale MJ发表综述:认为肿瘤恶液质情况下的身体组成变化与创伤,感染相似。1987 年Moley JF等认为肿瘤恶液质情况下的身体组成变化与厌食症也有显著不同,厌食症患者的体重丢失主要来源于脂肪丢失,极少数来源于肌肉。1992 年Fearon KC认为肿瘤恶液质患者的体重丢失相等地来源于肌肉及脂肪丢失。Fearon KC的这个讲座是1991年Sir David Cuthbertson奖章讲座。同样是他,10年后的2011年Fearon KC等专家发布了国际恶液质专家共识,将肿瘤恶液质定义为以骨骼肌块(skeletal muscle mass)持续(ongoing)下降为特征的多因素综合征,伴随或不伴随脂肪块(fat mass)减少,不能被常规的营养治疗逆转,最终导致进行性(progressive)功能障碍。1992年Fearon KC还认为:神经性厌食患者内在蛋白与肌肉蛋白成比例丢失,而肿瘤恶液质患者内脏蛋白是受保护的,甚至是升高的,丢失的蛋白质主要是肌肉蛋白质。
健康成年人的体重维持一个动态稳定,在一定时间、一定范围内的体重变化均属正常。体重变化幅度超过这一范围或体重变化的时间缩短均应视为异常。图4-3-2显示了正常成年人不同时间内的体重变化幅度。Bruera E 报告80%的上消化道肿瘤,60%的肺癌在第一次确诊时即有不同程度的体重丢失。体重丢失速度也是一个重要因素。1~2个月内体重丢失≥5%与6个以上体重丢失≥5%有显著不同的意义。
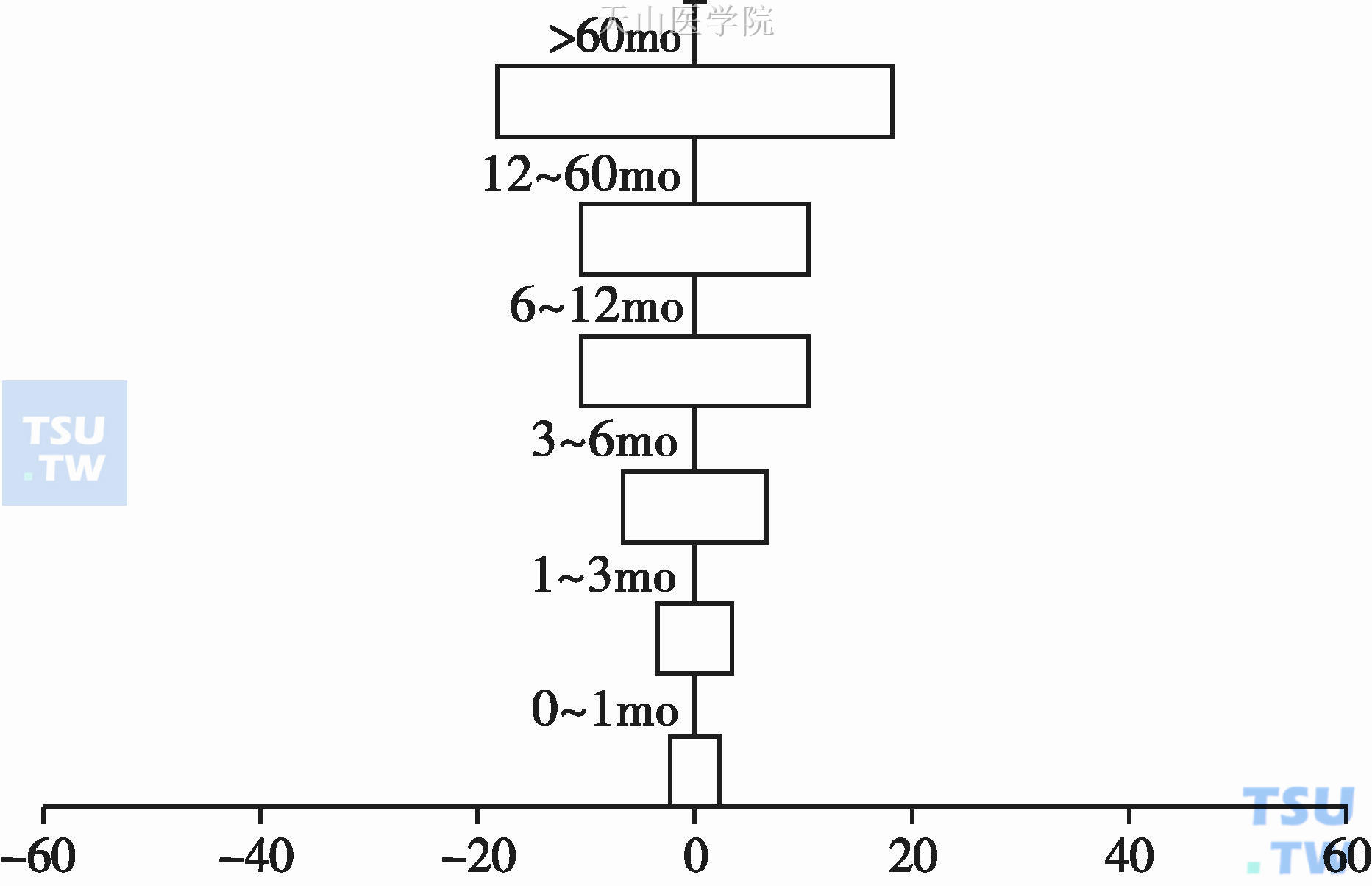
图4-3-2 健康成年人体重动态变化百分率(95%CI) mo,month,月
肿瘤恶液质患者体重丢失的同时,身体组成也在发生变化,身体组成的变化甚至可以发生在体重变化之前,体重稳定者也可能存在身体组成的变化。由此可见,肿瘤恶液质患者的体重丢失与身体组成的变化并非同步并行。肿瘤恶液质患者的身体组成变化的一个主要特征是瘦组织块的减少,伴随或不伴随脂肪块的减少。与单纯饥饿导致的身体组成变化显著不同,后者主要表现为脂肪块的减少,肌肉没有或很少减少;与创伤,感染时的身体组成变化也有显著差异,创伤、感染条件下的肌肉与脂肪同时减少。表4-3-1、表4-3-2比较了不同条件下体重丢失及身体组成的变化。
表4-3-1 不同条件下体重丢失及身体组成的变化
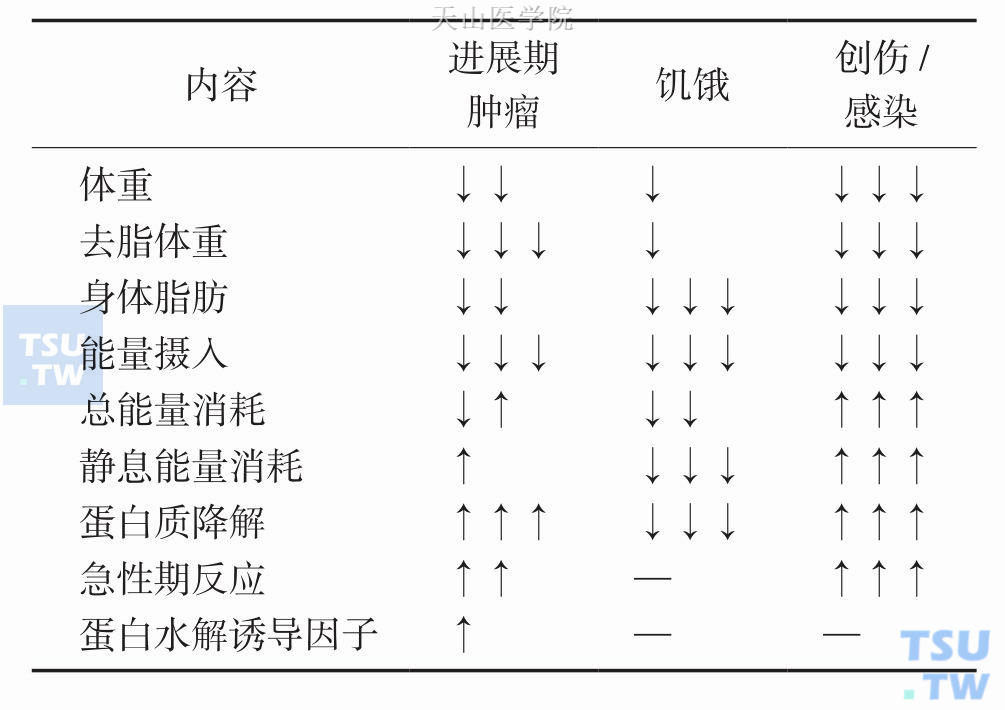
表4-3-2 标准健康人及肿瘤恶液质患者身体组成的比较
尽管厌食与恶液质常常结伴而行,但是,二者之间并没有原因与结果的关系。Bosaeus I 等观察了297例非选择性实体肿瘤患者,患者平均能量摄入为(26±10)kcal/kg,低于需要量,43%患者体重丢失>10%,48%的患者REE升高(>110%),代谢正常及代谢增高患者的食物摄入没有差异,肿瘤类型及性别与能量及蛋白质摄入也无关联,作者认为:体重丢失不能用食物摄入减少来解释,因为体重丢失患者与体重稳定患者的总能量摄入没有差别,体重丢失患者的每千克体重的平均能量摄入反而高于体重稳定的患者,食物摄入增加不能纠正体重丢失及代谢增强,他们认为肿瘤恶液质患者的体重丢失与摄入减少无关。
Uster A等则从另外一个角度说明了厌食与恶液质的关系。58例营养不足或高度营养不良风险的门诊肿瘤患者,随机分为两组,一组为常规治疗,另一组为标准营养干预(包括营养咨询,食品强化及口服营养补充),连续3个月,发现:与常规治疗组相比,营养干预显著提高了能量及蛋白质摄入量(能量+379kcal;95%CI,117~642;P=0.007;蛋白质+10.4g;95% CI,2.3~18.5;P=0.016)。但是,能量摄入的增加没有伴随体能状况及生活质量的改善。
尽管厌食不是体重丢失、特别是骨骼肌减少的直接原因,但却是肿瘤恶液质的一个重要组成成分。摄食是一项重要的社会互动,是一项患者家属可以参与并提供帮助的事情。
临床研究提示,营养支持不能逆转肿瘤患者的消耗进程,这些营养支持包括饮食咨询、TPN及各种食欲刺激剂的使用。而且,Evans WK等报告,肿瘤患者TPN治疗后体重增加是短暂的;TPN停止后,体重又将下降;身体组成分析提示增加的是脂肪组织及水分,而不是瘦体组织。相似的发现还见于HIV、创伤、危重病及脓毒症患者。
与肿瘤恶液质形成鲜明对照的是营养不良,抗肿瘤治疗引起的营养不良患者对营养补充治疗反应良好。而且,肿瘤放疗、化疗导致的营养不良对营养治疗也有很好的反应。这些患者接受营养师营养咨询后,体重丢失显著减少,营养状况改善,生活质量提高。胰腺肿瘤患者也是如此。虽然营养补充未能逆转体重丢失,但是却提高了生存率。还有人发现:高能量营养组患者比常规能量营养治疗患者成活得更长(50 vs. 32天)。
长期饥饿后,肝脏脂肪代谢产生的酮体替代葡萄糖成为大脑的能源物质,防止氨基酸糖异生,防止肌肉分解产生氨基酸。但是,恶液质情况下这种肌肉分解保护机制不存在,可能原因是肿瘤对宿主的能量需求非常大,足以阻止肝脏形成乙酰CoA,阻止乙酰CoA向乙酰乙酸及羟基丁酸转化,从而不能产生酮体。
恶液质时,脂肪组织丢失主要是由于脂解作用增强,因为与健康对照及无体重丢失的肿瘤患者相比,甘油及游离脂肪酸(free fatty acid,FFA)的转化升高。
与体重稳定的肿瘤患者相比,体重丢失者空腹血浆甘油、非酯化脂肪酸(nonesterified fatty acid,NEFA)及甘油三酯水平显著升高,他们对肾上腺素的脂解作用敏感性也升高。在业已完成TAG水解而没有再酯化的患者中,脂解作用增加40%,FFA氧化提高20%。恶液质患者对利钠肽(natriuretic peptide)的反应性也提高2~3倍,但是基础脂解率没有变化。抑制激素敏感脂肪酶(hormonesensitive lipase,HSL)可以削弱利钠肽的作用,恶液质患者脂肪细胞的HSLmRNA及蛋白质表达升高50%~100%,而总脂蛋白酯酶(lipoprotein lipase,LPL)及其mRNA无变化,尽管血浆甘油三酯及NEFA水平升高2倍。
脂肪酸(fatty acid,FA)以TAG形式储存于脂肪组织,构成90%的成年人能量储备。LPL从血浆脂蛋白中水解脂肪酸,并将脂肪酸输送到脂肪细胞里面,合成TAG。肾上腺素、胰高血糖素、促肾上腺皮质激素(adrenocorticotrophic hormone,ACTH),通过cAMP介导通路调节脂解。cAMP激活蛋白激酶(protein kinase,PKA),进而激活HSL,HSL是一分子TAG转化为三分子NEFA及一分子甘油这一过程的关键限速酶。Argilés JM 等认为HSL是肿瘤恶液质患者脂肪代谢中脂解增强的直接原因,是导致脂肪丢失的主要因素。
Fearon KC等报告摄入减少(<1500kcal/d)、体重丢失(≥10%)及系统炎症反应(CRP≥10mg/L)三者一起是预测体能状况及生存时间的有效变量,单独体重丢失不是预测预后的变量。
肌肉消耗
成年人肌肉块即使在没有刺激如运动情况下,也能保持稳定,所以蛋白质合成与蛋白质降解保持平衡。在肿瘤恶液质情况下骨骼肌萎缩,这种萎缩起源于蛋白质合成抑制,或蛋白质分解加强,或二者联合作用。对恶液质而言,究竟是蛋白质合成抑制还是蛋白质分解加强发挥更大的作用,目前有不同意见。Emery PW等亮氨酸标记示踪技术观察了肿瘤恶液质患者及志愿者蛋白质合成及蛋白质降解,发现肿瘤恶液质患者蛋白质合成显著减少,与对照组相比,差异显著(0.030 vs. 0.198%/h;P<0.01),而蛋白质合成及降解的整体速率没有明显变化。说明蛋白质合成抑制是肿瘤恶液质患者肌肉消耗的主要原因。他们建议,恶液质患者的治疗应该着眼于促进肌肉蛋白质合成。
Lundholm K等与Emery PW有类似的发现,他们观察了肿瘤恶液质患者,营养不良的非肿瘤患者,营养良好的危重患者及营养良好的择期手术患者四组患者血浆3-甲基组氨酸(3-methylhistidine)浓度,借以判断肿瘤恶液质患者大腿肌肉3-甲基组氨酸流出情况,发现营养良好的择期手术患者及危重患者,3-甲基组氨酸流出显著增多[(1.92±0.40)nmol/min/100g大腿组织,(0.93±0.32)nmol/min/100g大腿组织],而肿瘤恶液质患者及营养不良的非肿瘤患者其3-甲基组氨酸流出不显著。营养支持后,非肿瘤患者酪氨酸负平衡纠正,3-甲基组氨酸流出增加,而肿瘤患者酪氨酸仍然是负平衡,3-甲基组氨酸进一步减少。由于3-甲基组氨酸不能用于蛋白质合成,是蛋白质降解的直接证据,所以,作者认为肿瘤患者的体重丢失不是由于骨骼肌降解增加引起的,而是蛋白质合成抑制造成的。
但是,O'Keefe SJ等人有不同发现,他们用14C标记的亮氨酸观察整体蛋白质流(whole body protein flux)、蛋白质降解、合成及氧化率,发现慢性肝病及对照组患者内源性蛋白质降解及氧化相似,无显著差别;恶性肿瘤患者内源性蛋白质降解及氧化显著增强,肝细胞癌患者最高。肝癌患者蛋白质转化升高,氨基酸重掺入(reincorporation)蛋白质合成增加,肝癌患者肝脏活检提示肝脏蛋白质分数合成率升高。标记氨基酸掺入纤维蛋白原,免疫球蛋白G及转铁蛋白掺入率以肝癌患者最高。饮食调查发现,患者的摄食量为30%~70%需要量。他们认为肿瘤患者的骨骼肌丢失、蛋白质转化率升高主要来源于骨骼肌分解增强,以及骨骼肌分解游离氨基酸的氧化。尽管如此,更多的研究认为,肿瘤恶液质肌肉丢失是蛋白质合成抑制及蛋白质降解增强两者共同作用的结果。肌肉块的减少与肿瘤不良临床结局密切相关。Wallengren O等观察了471例进展期患者最后2年的肌肉块变化及其与年龄、性别、肿瘤类型、炎症的关系,发现:年龄>71岁的患者肌肉块明显减少[(-1.1±0.3)kg,P<0.001],肌肉块减少的发病率男性高于女性 (59%vs. 28%,P<0.001),男性随着时间的推移肌肉块显著减少[(平均1.4±0.3) kg/年,P<0.001],而女性无明显变化[(0.3±0.4) kg/年,P=0.5],胰腺癌患者肌肉块丢失比胆管癌、结直肠癌患者明显 (P<0.02),不同年龄、不同肿瘤类型患者随着时间推移其肌肉块变化没有显著的组间差异,61%~70%的患者在研究过程中其CRP升高,CRP>10mg/L者其肌肉块更少[(0.6 ± 0.2) kg,P<0.001]、并且在疾病进程中加速丢失肌肉块[(0.7±0.3) kg/年,P=0.03]。作者认为:肿瘤患者的肌肉块减少与年龄、性别、肿瘤类型及炎症有关。图4-3-3
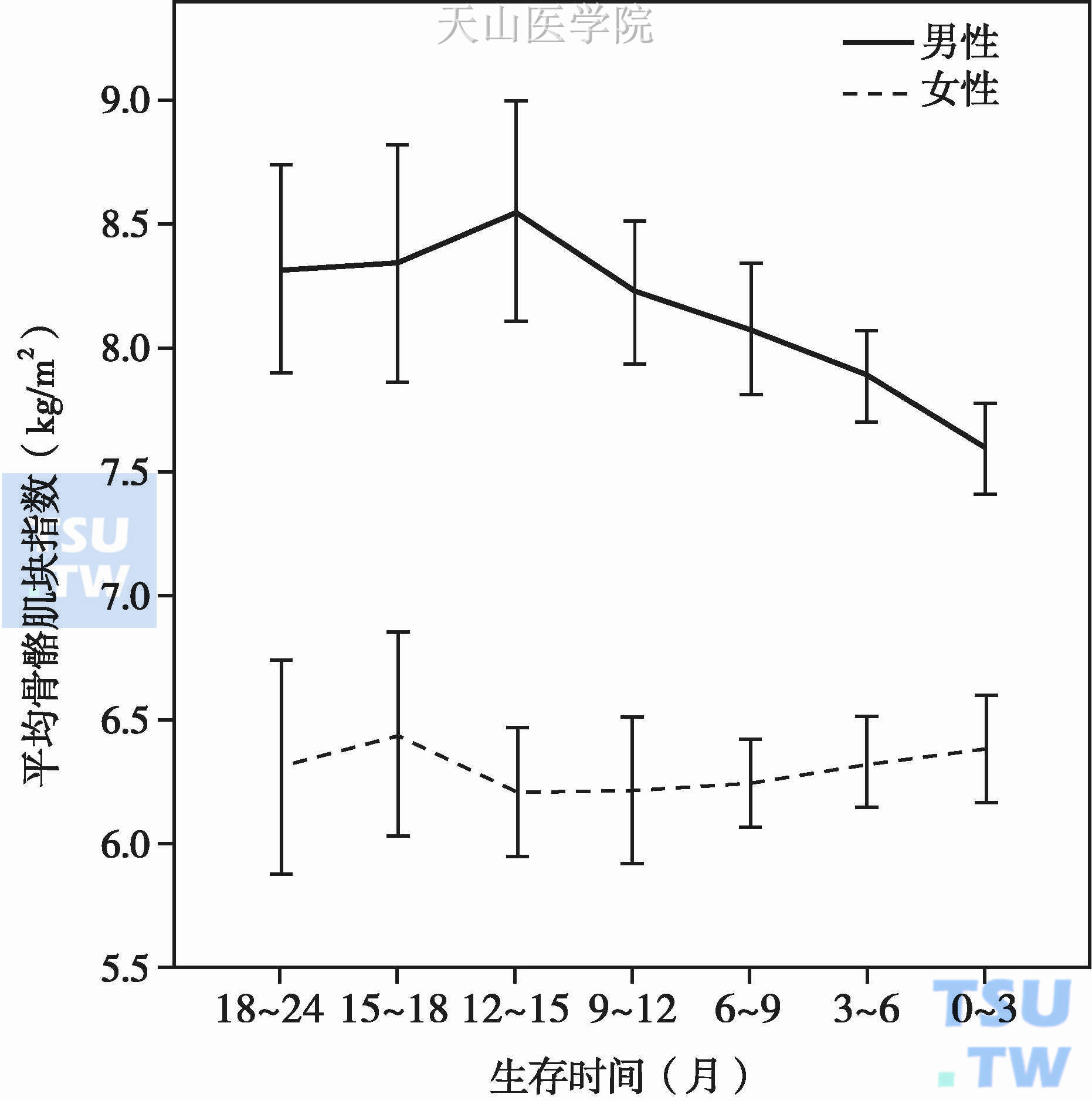
图4-3-3 进展期肿瘤患者肌肉块的动态变化
肿瘤恶液质时,含快缩Ⅱ型纤维的肌肉如胫骨前肌,腓肠肌丢失快于含慢缩Ⅰ型纤维的肌肉如比目鱼肌,因为Ⅱ型纤维对恶液质性刺激反应更敏感,致使蛋白质氧化增强,蛋白质表达抑制。Ⅱ型纤维中抗氧化剂基因,一氧化氮(nitric oxide,NO)及iNOS减少。此外,肌球蛋白同工型在Ⅰ型纤维表达降低,在Ⅱ型纤维表达升高。
研究发现,肿瘤恶液质肌肉消耗还与抗肌萎缩蛋白-糖蛋白复合物(dystrophin-glycoprotein complex,DGC) 有关。DGC是一种与肌肉营养不良有关的膜结构,胃肠道肿瘤患者其抗肌萎缩蛋白(dystrophin)水平降低,而DGC糖基化增强。还有研究发现,抗肌萎缩蛋白缺如小鼠肌肉消耗显著,而抗肌萎缩蛋白转基因小鼠肌肉消耗减少。所有这些均提示DGC功能障碍在肿瘤恶液质发病机制中扮演重要角色。
肿瘤恶液质的最重要特征之一是肌肉蛋白质代谢异常,肌肉蛋白质合成代谢与分解代谢不平衡,分解代谢大于合成代谢,最终导致肌肉消耗。研究发现,所有的恶性肿瘤都存在肌肉蛋白质代谢异常,只有程度的差异,速度快慢的差异。肌肉蛋白质分解率升高,新蛋白质合成率下降,导致净蛋白分解(net protein breakdown)。肿瘤患者蛋白质转化(protein turnover)升高,与伴有相同程度体重丢失的良性疾病相比,差异非常显著,导致能量需求增加约100kcal/d。恶液质患者从骨骼肌氧化BCAA增强,以供糖异生之用。BCAA氧化增多,使糖异生底物增多,糖异生进一步激活,进而引起肌肉进一步降解,能量消耗进一步增加。BCAA除了作为骨骼肌蛋白质合成的底物外,BCAA还有刺激蛋白质合成的特殊作用,BCAA通过启动信号传导通路,进而启动蛋白质翻译。在BCAA中,亮氨基酸的作用最为强大。研究发现它可以抑制MAC16接种小鼠的肌肉丢失,促进蛋白质合成,抑制蛋白质降解,从而增加肌肉块重量。
一共有三条蛋白质水解通路参与肿瘤恶液质肌肉消耗:①溶酶体系统,包括半光氨酸蛋白酶cathepsins B、 H、L及天冬氨酸蛋白酶cathepsin D,该通路主要负责细胞外蛋白质及细胞受体的降解;②钙激活系统,包括钙激活酶(calpains)Ⅰ、Ⅱ,主要参与组织损伤,坏死及自溶;③泛素-蛋白酶体通路(ubiquitin-proteasome pathway,UPP),它需要ATP,并与calpain系统协同工作,分解并降解肌丝。肿瘤动物及肿瘤患者的研究均提示,UPP系统在肌肉纤维蛋白降解中发挥主要作用,对体重丢失>10%的患者而言UPP作用更大。转录因子Foxo3控制肌肉UPP及溶酶体系统,但是机制不同。体重丢失较少(2.9%)的患者可能主要是溶酶体系统发挥主要,而体重丢失较多(>10%)的患者UPP发挥主要作用。因为在体重丢失较少的患者活检中没有发现UPP的变化(升高),而cathepsinB mRNA表达显著增加,而溶酶体本身没有参与骨骼肌纤维蛋白降解。
