
垂体或下丘脑的多种病损可累及腺垂体的内分泌功能,当垂体的全部或绝大部分被毁坏后,产生一系列内分泌腺功能减退表现,主要累及的靶腺为性腺、甲状腺及肾上腺皮质,临床上称为腺垂体功能减退症。成人腺垂体功能减退症又称为Simmond病。最常见的病因为产后垂体缺血性坏死(由Harold Leeming Sheehan首次报道,故亦称为Sheehan综合征)及垂体瘤。按发病部位和病因,可将腺垂体功能减退症分为原发性腺垂体功能减退症(primary hypopituitarism)和继发性腺垂体功能减退症(secondary hypopituitarism)两类,前者又可分为先天性(遗传性)腺垂体功能减退症和获得性腺垂体功能减退症两种。
腺垂体功能减退症多见于成年(21~40岁)女性,其病因有:①垂体病变或发育异常致腺垂体激素分泌减少;②下丘脑病变导致腺垂体激素释放激素分泌不足;③下丘脑-垂体的联系(垂体门脉系统)中断,下丘脑促腺垂体激素不能到达腺垂体而引起功能减退。
腺垂体功能减退的病因

血管病变造成垂体坏死
血管病变造成的垂体坏死分为产后垂体坏死(postpartum pituitary necrosis)和非产后垂体坏死两种。
产后缺血性垂体坏死
为女性腺垂体功能减退的常见病因,发病机制未明,临床主要分为2种情况。
一、分娩大出血或其他并发症
容易造成垂体坏死与妊娠及分娩时垂体的变化有关,分娩后,垂体增生肥大的因素突然消失,垂体迅速复旧,腺垂体血流量减少。在此种情况下发生周围循环衰竭,腺垂体因缺血而坏死(Sheehan综合征)。由于神经垂体的血液供应不依靠垂体门脉系统,一般不被累及,但如果缺血严重而持久可同时累及神经垂体而并发尿崩症。分娩时大出血与循环虚脱使腺垂体动脉痉挛而闭塞。这些动脉除有少数分支直接供应垂体外,在垂体柄周围分成微血管,后者进入下丘脑正中隆突和垂体柄,与该处的神经分泌纤维紧密相接,便于下丘脑神经激素进入微血管内;微血管再汇合成垂体门脉系统供应腺垂体。动脉闭塞后,垂体门脉血源供应断绝,而动脉痉挛可能与休克时交感神经兴奋或与使用麦角碱、AVP或其他缩血管药物有关。
随着现代产科技术的进步,典型而严重的Sheehan综合征已经少见,代之以非典型的轻度Sheehan综合征,而且可以在分娩后数年才发病,如果能排除原发性甲减、甲旁减、糖尿病、多囊卵巢综合征等疾病,那么毛发脱落、乏力、月经稀少或闭经的最大可能是分娩事件遗留下来的腺垂体功能减退症。
二、弥散性血管内凝血
子痫、胎盘早期剥离、羊水栓塞、感染性休克可引起弥散性血管内凝血(disseminated intravascular coagulation,DIC)。垂体最常成为DIC的损伤组织而导致坏死,血液循环不易恢复与前述的垂体迅速复旧,血流量减少以及腺垂体血液供应主要依靠垂体门脉而非动脉有关。
产后非缺血性垂体坏死
除产后垂体缺血性坏死外,其他血管病变亦可成为腺垂体功能减退的病因,如糖尿病大血管和微血管病变、海绵窦血栓形成、颞动脉炎、颈动脉瘤、抗中性粒细胞胞浆抗体(antineutrophil cytoplasmic antibody,ANCA)性血管炎等。
颅内病变和全身疾病引起非产后腺垂体功能减退症
颅内肿瘤
为引起腺垂体功能减退症的另一重要病因,成年人最常见者为垂体瘤,儿童最常见者为颅咽管瘤。其他下丘脑-垂体瘤的扩张性生长、卒中或压迫垂体也可引起本症。
空泡蝶鞍综合征
常并发腺垂体功能减退;发生垂体功能减退后,空泡蝶鞍的程度常进一步加重,两者互为因果。
颅脑创伤或垂体柄离断
严重颅脑创伤是腺垂体功能减退症的重要原因,称为“颅脑创伤后腺垂体功能减退症(head posttraumatic hypopituitarism)”。据报道,28%~69%的颅脑创伤患者可并发程度不等的腺垂体功能减退症,且在急性期伴有高钠血症或低钠血症(AVP缺乏或AVP分泌过多)。腺垂体功能减退症可在颅脑创伤的急性期、恢复期或在颅脑创伤数月至数年后发病。颅脑创伤可引起急性腺垂体大片梗死,或由于垂体柄折断及垂体门脉中断而发病,病变累及垂体柄者常同时并发尿崩症。这些患者大多因颅骨骨折累及颅底或垂体窝,垂体坏死使预后更为严重。幸存者在遇有各种应激时,常发生垂体危象(pituitary crisis)。垂体手术亦可引起腺垂体功能减退,近年开展的立体定位放射手术(stereotactic radiosurgery)已经使该并发症明显下降。
垂体柄离断引起垂体柄离断(断裂)综合征(pituitary stalk interruption syndrome)导致永久性GH缺乏和血清葛瑞林明显升高(下丘脑损害的结果)。因而,高葛瑞林血症可作为垂体柄离断综合征的诊断线索和依据之一。
头颈部放射治疗
下丘脑-垂体卒中对放射性特别敏感,头颈部放射治疗后数年常出现腺垂体功能减退症。由于下丘脑受损,而腺垂体功能减退是继发性的,TRH兴奋后,血TSH峰值延迟。放射线通过离子化肿瘤细胞的DNA而发挥治疗作用。放射线破坏细胞DNA的空间结构或产生自由基,导致细胞死亡,离子型放射线也引起正常神经细胞和神经胶质细胞变性、脱髓鞘和死亡;由于血管变性与内皮细胞死亡改变了血管通透性,引起组织水肿和纤维化。放射治疗所致的下丘脑-垂体损伤可分为早发性和迟发性两类,部分表现为全垂体功能减退,多数表现为GH缺乏(32%)、LH/FSH缺乏(27%)、ACTH缺乏(21%)或TSH缺乏(9%)。
放射性垂体损伤的另一种结局是青春期发育异常,一般可有以下几种表现:①青春期发育提前:属于中枢性性早熟的表现之一;②青春期发育加速(rapid puberty):其特点是青春期发育的启动年龄基本正常,但发育和进展的速度加快,女性从乳腺开始发育到月经初潮的时间短于18个月,而男性的睾丸体积在1年内达到10ml以上;③线性生长减慢:青春期发育的启动时间正常,但发育的进展缓慢,其最终身高一般低于预计高度;④青春期发育延迟:放射性损害较重,这些患者除了青春期发育延迟外,以后往往发生低促性腺激素性性腺功能减退症;⑤青春期发育停止(arrested puberty):⑥青春期发育的启动时间正常或甚至提前,但发育到达一定阶段后不再进展,这些患者常并发永久性GHD。
成年女性患者接受大剂量放射治疗后,可并发月经紊乱、继发性闭经、卵巢功能减退、骨质疏松或卵巢早衰;男性患者多出现性欲减退、阴茎勃起障碍、精子数目下降、睾酮水平降低、肌肉消瘦和骨质疏松。放射治疗还可以引起ACTH、TSH、GH缺乏及高泌乳素血症,出现相应的临床表现。
自身免疫性垂体损害
垂体自身免疫性损害可能存在多种临床情况,有些自身免疫性垂体损伤是全身疾病在垂体的表现。
- 淋巴细胞性垂体炎:淋巴细胞性垂体炎(lymphocytic hypophysitis)为垂体的自身免疫性炎症性疾病,部分病例与其他自身免疫性疾病并存。可见于任何年龄,以围分娩期女性多见。而非妊娠妇女和男性很少发病。典型者的表现为头痛、突眼(海绵窦炎)、尿崩症、视野缺损和垂体功能减退(ACTH和TSH缺乏,LH、FSH和GH多正常),有时可伴有淋巴细胞性甲状腺炎、Addison病或恶性贫血。MRI有较特异表现,有些患者糖皮质激素治疗无效;MRI显示垂体均匀性增大而肿瘤不典型或伴有自身免疫性疾病时,要注意排除淋巴细胞性垂体炎可能。
- 免疫球蛋白G4相关性系统性疾病:免疫球蛋白G4相关性系统性疾病(immunoglobulin(Ig)G4-related systemic disease,IgG4 RSD)是近年来新确立的一种自身免疫性疾病,多见于中老年男性;IgG4 RSD的主要表现为自身免疫性胰腺炎,可伴有唾液腺、肺、肝、胆管、肾、腹膜后等组织的自身免疫性炎症。常合并有漏斗-垂体炎(infundibulo-hypophysitis)和尿崩症。垂体肥大伴有垂体柄增厚,部分患者合并有肥厚性硬脑膜炎(hypertrophic pachymeningitis)和鼻窦炎(sinusitis)。患者对糖皮质激素的反应良好。血清免疫球蛋白G4明显升高,受累组织有明显IgG4阳性浆细胞浸润。采用补充/替代剂量的糖皮质激素(氢化可的松)对部分患者有效。
- 新生儿肝炎:为先天性腺垂体功能减退的病因之一,主要表现为甲减和肾上腺皮质功能减退,经补充/替代治疗后,新生儿肝炎可逐渐恢复正常。在临床上,对久治不愈的新生儿肝炎患者要考虑并发腺垂体功能减退症可能。
- 产后自身免疫性垂体炎:近年发现,有些产妇并无分娩困难、出血、昏厥或感染史,但产后无乳汁分泌,并常伴有厌食、嗜睡、性欲减退、活动能力低下等症状,有的患者以贫血为主要表现。经垂体功能的动态试验证实有轻度腺垂体功能减退症,称为产后自身免疫性垂体炎(postpartum autoimmune hypopituitarism),其病因未明,可能与垂体自身免疫性病变有关。另一种情况是产后自身免疫性垂体炎合并产后甲状腺炎(postpartum thyroiditis),两种疾病可同时或先后发作,提示两者在病因上存在联系。
- 特发性腺垂体功能减退症:部分腺垂体功能减退患者无明确病因可查,一般认为是由于某种自身免疫反应导致垂体退化萎缩所致。
垂体感染
微生物感染可通过不同方式使腺垂体受损,垂体脓肿可直接毁坏垂体,颅底脑膜炎可影响下丘脑激素的转运,脑炎可影响下丘脑神经激素的合成与分泌。严重全身性感染(如伤寒)也可引起本症。
全身性疾病
可引起腺垂体功能减退症的全身性疾病很多,常见的有:
- 浸润性病变(如白血病、淋巴瘤、黄色瘤、结节病、血色病等),其中结节病可有广泛下丘脑浸润,而垂体本身并无明显损害;
- 严重营养不良(如神经性厌食、胃切除或蛋白-热卡营养不良症);
- 长期而严重的神经症可伴有轻度腺垂体功能减退症;
- 可卡因相关性垂体功能减退症为血管病变的一种表现,其特点是全垂体功能减退症伴或不伴尿崩症;
- 糖尿病。
下丘脑-腺垂体畸形/发育不良导致腺垂体功能减退
在垂体的胚胎发育过程中,需要许多控制细胞发育的因子参与调节。如果相关的发育因子缺乏即可引起垂体激素缺乏性垂体功能减退症。主要有两种类型,一是调节垂体发育的基因突变或缺失引起腺垂体发育不良和腺垂体激素分泌不足;二是先天性畸形累及下丘脑-垂体。先天性腺垂体发育或结构异常主要见于:①垂体发育不良与垂体不发育;②先天性腺垂体功能减退症伴神经垂体异位(congenital hypopituitarism with posterior pituitary ectopia)可能是先天性腺垂体功能减退症的一种特殊类型。腺垂体功能减退为部分性,多为GH和(或)ACTH缺乏;尿崩症为中枢性。Lukezic等报道15例年龄为13~38岁先天性腺垂体功能减退症伴神经垂体异位患者(男12例,女3例),其中11例有多种垂体激素分泌缺陷,4例为单一性GHD,无一例患者有口干、多饮和多尿。5例高渗盐水试验异常,说明这些患者中有相当部分患者伴有亚临床AVP分泌障碍和渴感功能的减退。
脑中线-视神经发育不良症
脑中线-视神经发育不良症(septo-optic dysplasia,SOD)亦称de Morsier 综合征,属于罕见的先天性发育异常性疾病。临床上以脑中线发育异常(midline abnormalities in central nervous system)、视神经发育不良(optic nerve hypoplasia)和下丘脑-垂体发育障碍三联征为特征。该综合征首次报道于1941年,发病率占活婴的1/10 000。
多数SOD病例为散发性,家族性者少见。近年发现,同源框基因(homeobox gene)Hesx1和SOX2突变产生SOD表型,HESX1(OMIM 601802)基因纯合子突变(常染色体隐性遗传)或杂合子突变(常染色体显性遗传)。SOX2突变引起下丘脑性低促性腺激素性性腺功能减退症(GnRH缺乏所致)和眼发育障碍(如无眼、小眼等),即无眼-食管闭锁-生殖器异常(anophthalmia-esophageal atresia-genital anomalies)综合征,见下表。目前鉴定了13个不同的突变位点,导致胚胎第4~6周前神经板(anterior neural plate)形态发生和前脑发育不良。但是,Hesx1和SOX2突变仅能解释小部分病例的病因,其他相关基因突变有待研究。
HESX1与SOX2 突变引起的垂体功能异常
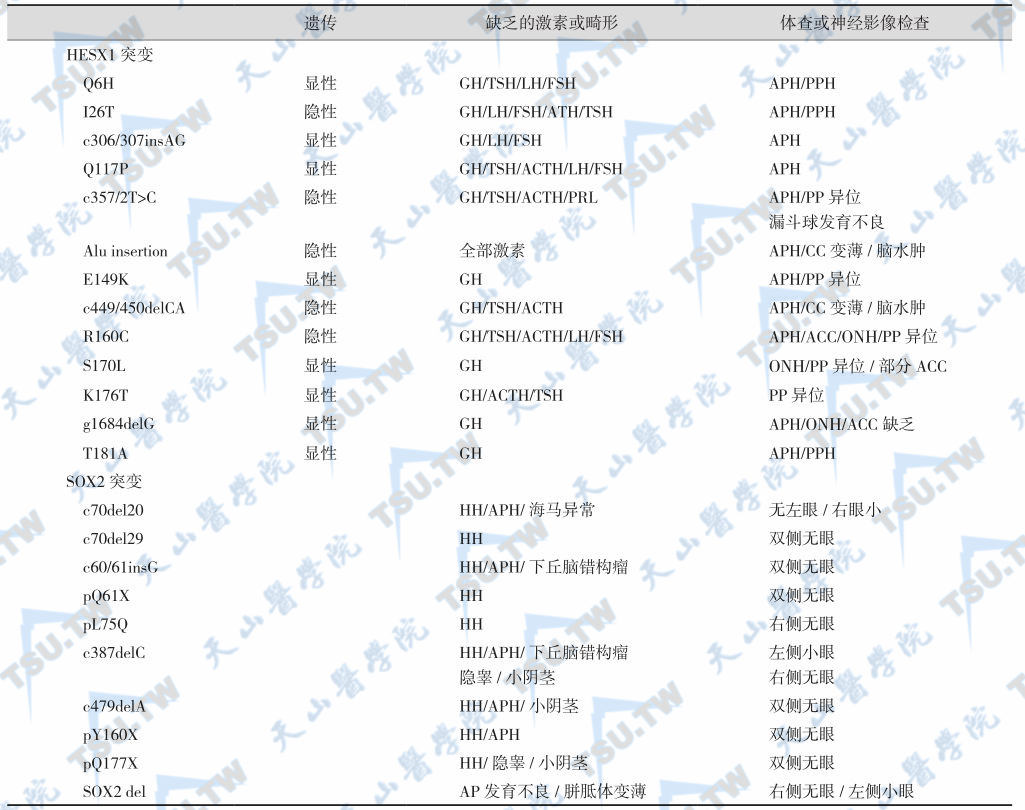
注:ACC:agenesis of the corpus callosum,胼胝体不发育;AP:anterior pituitary,腺垂体;APH:anterior pituitary hypoplasia,腺垂体发育不良;HH:hypogonadotropic hypogonadism,低促性腺激素性性腺功能减退症;ON:optic nerve,视神经;ONH:optic nerve hypoplasia,视神经发育不良;PP:posterior pituitary,神经垂体;PPH:posterior pituitary hypoplasia,神经垂体发育不良
视神经发育不良是患者就诊的主要原因,发生率约70%,突出表现是视力减退和眼球震颤。中枢神经中线异常主要包括胼胝体不发育、小脑发育不全、脑裂、透明隔缺如等。MRI可见脑组织形态异常、部分脑结构缺乏和神经垂体异位(75%~80%)。检查可见脑电图异常,严重患者可能存在癫痫、偏瘫、智力障碍等。临床表现具有显著的异质性,主要包括双侧视神经发育不良、眼球震颤、躯体发育延迟、神经和下丘脑-垂体发育障碍(如垂体细小神经垂体异位、垂体柄变薄或缺乏、空泡蝶鞍等),垂体激素缺乏较常见,是先天性垂体功能减退的最主要原因。垂体激素缺乏相当常见(62%~80%),按照发生频率依次为GH缺乏(85%)、ACTH缺乏(48%)、TSH缺乏(33%)、LH/FSH缺乏(8%)。儿童期出现者症状重,偶尔发生垂体功能减退症危象。但一般症状不明显,多数需要用动态试验才能确诊。
其他临床综合征
除HESX1与SOX2外,引起脑结构发育障碍、躯体畸形和复合型垂体激素缺乏(combined pituitary hormone deficiency,CPHD)的发育因子(developmental factors)还有LHX3(颈项短小与强直,short stiff neck)、LHX4(小脑发育异常,cerebellar anomalies)、OTX2(小眼-无眼,microphthalmia and anophthalmia)、SOX3(智力障碍,mental retardation)、TBCE(甲旁减-精神迟钝-同质异型,hypoparathyroidism-,retardation and dysmorphism)、PITX2、pit1、PROP1、XIX6、POU1F1、TBX19等。引起单基因表达的发育因子有GH1、GHRHR、GnRHR1、TBX19(TPIT)、PMOC、TSHβ、HRHR、LHβ/FSHβ等。这些发育因子突变导致单一垂体激素缺乏,而其他激素的合成与分泌正常。此外,约半数患者伴有神经垂体异位。少数病例存在两种或更多的致病因素。
- 发育调节因子突变:主要见于:①KAL-1突变(FSH 和LH缺乏);②Laurence-Moon综合征(FSH和LH缺乏);③Prader-Willi综合征(FSH和LH缺乏)。
- 激素受体突变:主要有:①ACTH受体(ACTH缺乏);②GHRH受体(GHD);③GnRH受体(FSH和LH缺乏症);④瘦素受体和黑皮素受体(LH和FSH缺乏症)。
- 转录因子突变:参与垂体发育的转录因子很多,这些转录因子突变可导致腺垂体功能减退症:①pit-1基因突变:导致先天性GH、PRL和TSH缺乏,其特点是垂体正常或萎缩;②腺垂体特异性配对的同型结构域转录因子-1(Prop-1)基因突变:引起多种腺垂体激素(GH、TSH、PRL、LH和FSH)缺乏,MRI可发现蝶鞍高度下降、容积缩小(或正常),有时明显增大或伴神经垂体异位;③ DAX1突变:导致肾上腺发育不良和LH与FSH缺乏;④ T-Pit突变:引起ACTH缺乏所致的继发性肾上腺皮质功能减退症,患者伴有肥胖,毛发可变为红色;⑤秃顶-神经缺陷-内分泌病综合征(ANE综合征,alopecia-neurological defects-endocrinopathysyndrome)是新近鉴定的一种常染色体隐性遗传性综合征,病因与核糖体合成相关蛋白RBM28突变有关。除男性乳腺发育外,主要表现为复合型垂体激素(GH、LH、FSH、ACTH 、TSH、PRL 等)缺乏(combined pituitary hormone deficiency,CPHD),其他临床特征包括皮肤损害(秃顶、面部多发性皮肤痣、皮肤囊肿、皮肤色素沉着)、先天性神经发育缺陷、智力障碍、进展型运动功能障碍、牙齿异常。
